
In Teil eins dieses ArtikelsIch habe den vertraglichen und regulatorischen Rahmen überprüft, den die US-Regierung für die anfängliche Entwicklung, Herstellung und den Erwerb der Covid-mRNA-Impfungen anwendet, und habe dabei die BioNTech/Pfizer-Vereinbarungen zur Veranschaulichung des Prozesses herangezogen.
Ich habe nachgewiesen, dass diesen Produkten auf der Grundlage klinischer Studien und Herstellungsverfahren eine Notfallzulassung (EUA) erteilt wurde
- keine verbindlichen Rechtsnormen,
- keine gesetzlich vorgeschriebene Sicherheitsaufsicht oder -regelung, und
- Es besteht kein Rechtsanspruch des Herstellers für mögliche Schäden.
In diesem Folgeartikel werde ich eine detaillierte Analyse der zugrunde liegenden Dokumentation liefern.
Andere Transaktionsautorität/-vereinbarung (OTA): Ein Weg zur militärischen Akquisition
Das Vereinbarung Der Vertrag zwischen der US-Regierung, vertreten durch das Verteidigungsministerium (DoD), und Pfizer, Vertreter der BioNTech/Pfizer-Partnerschaft, im Juli 2020 über den Kauf eines „Impfstoffs zur Vorbeugung von COVID-19“ war kein gewöhnlicher Kaufvertrag.
Es handelte sich um eine Vereinbarung im Rahmen der Other Transaction Authority (OTA) – ein Übernahmeweg, der laut Richtlinien des Verteidigungsministeriums, wird seit 1958 verwendet, um „einer Bundesbehörde den Eintritt zu gestatten andere Transaktionen als Verträge, Zuschüsse oder Kooperationsvereinbarungen"
[Fettschrift hinzugefügt]
Eine ausführliche Übersicht über die Nutzung von OTA durch das Verteidigungsministerium, einschließlich seiner gesetzlichen Geschichte, finden Sie im Bericht des Congressional Research Service vom 22. Februar 2019. In diesem Bericht und in allen anderen Diskussionen über OTA wird darauf hingewiesen, dass es sich dabei um einen alternativen Erwerbsweg handelt für Verteidigungs- und Militärzwecke. Es ist nicht für irgendetwas gedacht, das in erster Linie für den zivilen Gebrauch bestimmt ist, und wurde vor Covid auch nie verwendet.
Wenn Sie danach suchen OTA-Gesetze im US-Kodex, das ist der Weg, den Sie gehen werden:
Streitkräfte -> Allgemeines Militärrecht -> Beschaffung -> Forschung und Technik -> Vereinbarungen -> Befugnis des Verteidigungsministeriums zur Durchführung bestimmter Prototypenprojekte
Dieser rechtliche Weg zeigt sehr deutlich, dass OTA-Gesetze für den Erwerb von Forschungs- und Technikprototypen für die Streitkräfte gedacht sind.
Das Verteidigungsministerium ist für drei verschiedene Arten von OTs zuständig: (1) Forschungs-OTs, (2) Prototyp-OTs und (3) Produktions-OTs.
Diese drei Arten von OTs repräsentieren drei Phasen der anfänglichen Forschung, der Entwicklung eines Prototyps und der schließlichen Produktion.
Innerhalb dieser drei Arten gibt es bestimmte Kategorien von Projekten, für die sich OTA bewerben kann:
- Ursprünglich laut der OTA-Übersicht Nach Angaben des Verteidigungsministeriums war die Other Transaction Authority „auf Waffen oder Waffensysteme beschränkt, deren Erwerb oder Entwicklung durch das Verteidigungsministerium vorgeschlagen wurde“.
- OTA wurde später erweitert und umfasste „jedes Prototypprojekt, das in direktem Zusammenhang mit der Verbesserung der Missionseffektivität des Militärpersonals und der unterstützenden Plattformen, Systeme, Komponenten oder Materialien steht, die vom Verteidigungsministerium erworben oder entwickelt werden sollen, oder mit der Verbesserung von Plattformen, Systemen und Komponenten.“ oder Materialien, die von den Streitkräften verwendet werden.“
Bisher klingt nichts davon nach einem Erwerbsweg für Millionen neuartiger Medizinprodukte, die hauptsächlich für den zivilen Gebrauch bestimmt sind.
Gibt es eine Ausnahme für die zivile Nutzung von OTA, die möglicherweise für Covid-mRNA-Impfstoffe gilt?
Das GJ2004 National Defense Authorization Act (P.L. 108-136) enthielt einen Abschnitt, der „dem Leiter einer Exekutivbehörde, die sich mit Grundlagenforschung, angewandter Forschung, fortgeschrittener Forschung und Entwicklungsprojekten befasst“ die Befugnis zu anderen Transaktionen einräumte, „die das Potenzial haben, die Verteidigung gegen oder die Erholung von Terrorismus oder nuklearen, biologischen, chemischer oder radiologischer Angriff.“
Diese Bestimmung wurde bis 2018 verlängert, scheint jedoch nicht über dieses Jahr hinaus verlängert worden zu sein. Beachten Sie außerdem, dass selbst in diesem Ausnahmefall der Nicht-DoD-Nutzung von OTA Es muss sich um Terrorismus oder einen Angriff mit Massenvernichtungswaffen (CBRN) handeln..
Welche anderen OTA-Gesetze könnten gelten?
Der oben zitierte CRS-Bericht 2019 enthält dieses Diagramm, aus dem hervorgeht, dass einige Nicht-DoD-Agenturen über OTA- oder verwandte Behörden verfügen:
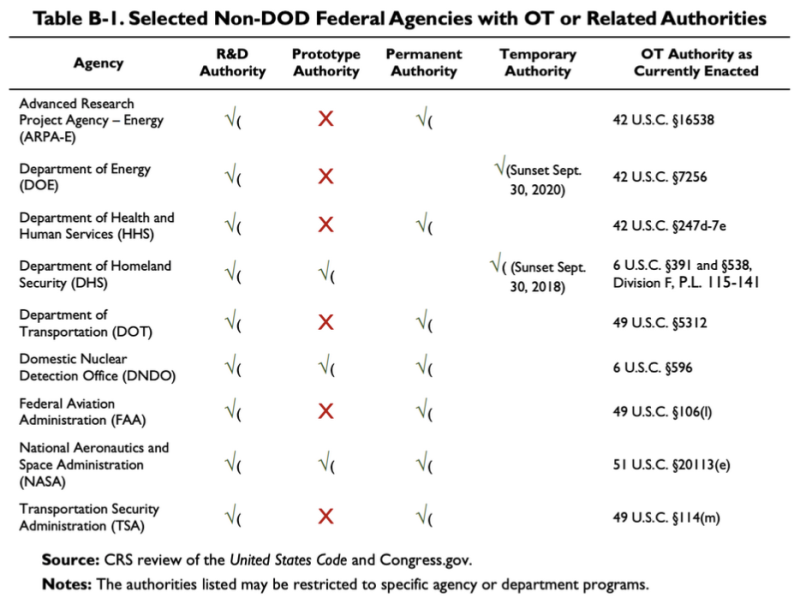
Laut dieser Tabelle verfügt das Ministerium für Gesundheit und menschliche Dienste (HHS) über einige Forschungs- und Entwicklungsbehörden (F&E) und andere Transaktionsbehörden. Das Gesetz über die Die OT-Behörde des HHS ist 42 U.S.C. §247d-7e.
Wo ist dieses Gesetz untergebracht und was steht darin?
Öffentliche Gesundheit und Wohlfahrt -> Öffentlicher Gesundheitsdienst -> Allgemeine Befugnisse und Pflichten -> Zusammenarbeit zwischen Bund und Ländern -> Biomedical Advanced Research and Development Authority (BARDA) -> Transaktionsbehörden
Es gibt also eine Stelle im Gesetz über die Gesundheit und das Wohlergehen der Zivilbevölkerung, an der OTA anwendbar sein könnte, obwohl sie gültig ist Nur für Forschung und Entwicklung, nicht für Prototypen oder Fertigung.
Das Gesetz besagt, dass der BARDA-Sekretär über OT-Autorität verfügt
in Bezug auf ein Produkt, das ein Produkt ist oder werden könnte qualifizierte Gegenmaßnahme oder eine qualifiziertes Pandemie- oder Epidemieprodukt, Aktivitäten, die überwiegend—
(i) nach Grundlagenforschung und präklinischer Entwicklung des Produkts durchgeführt werden; Und
(ii) im Zusammenhang mit der Herstellung des Produkts im kommerziellen Maßstab stehen und in einer Form, die den regulatorischen Anforderungen des Bundes entspricht Lebensmittel-, Arzneimittel- und Kosmetikgesetz [21 USC 301 et seq.] oder darunter Abschnitt 262 dieses Titels.
[Fettschrift hinzugefügt]
Die im Gesetz aufgeführten „regulatorischen Anforderungen“ bedeuten, dass es für BARDA/HHS unmöglich wäre, Vereinbarungen – auch nur im Bereich Forschung und Entwicklung – für medizinische Produkte (wie die mRNA-Impfstoffe) abzuschließen, die keinen strengen Sicherheitstests und einer strengen Herstellungsaufsicht unterzogen wurden.
HHS-„Partnerschaft“ mit DoD umging Zivilschutzgesetze
Um die missliche Lage anderer Transaktionsautoritäten/-vereinbarungen in Bezug auf zivile Behörden im Allgemeinen und Covid-mRNA-Impfstoffe im Besonderen zusammenzufassen:
- OTA wurde geschrieben und kodifiziert, um dem Militär die Möglichkeit zu geben, ohne großen bürokratischen Aufwand Waffen und andere notwendige Systeme und Ausrüstung zu erwerben. Es umfasst Forschung und Entwicklung, Prototypen und die anschließende Fertigung.
- Der einzige OTA für eine öffentliche Gesundheitsbehörde ist für das HHS und deckt nur Forschung und Entwicklung ab, nicht Prototypen oder Fertigung.
- Sogar die dem HHS erteilte OTA für Forschung und Entwicklung verlangt immer noch, dass Produkte „in einer Form hergestellt werden, die die regulatorischen Anforderungen“ für die Arzneimittel- und Impfstoffsicherheit erfüllt.
Mit anderen Worten: HHS hätte seine sehr begrenzte OTA auf keinen Fall nutzen können, um Verträge für Hunderte Millionen neuartiger medizinischer Produkte zu unterzeichnen.
Was hat HHS also getan?
Wie das Government Accountability Office (GAO) in seinem Bericht feststellte Bericht vom Juli 2021 zum Thema „Covid-19 Contracting:“ HHS „arbeitete“ mit dem Verteidigungsministerium zusammen, um „die OTA-Behörden des Verteidigungsministeriums zu nutzen … die dem Verteidigungsministerium fehlten“. (S. 24)
Was sind die OT-Behörden des Verteidigungsministeriums für Medizinprodukte?
Wie bereits erwähnt, soll OTA dem Militär helfen, ohne großen bürokratischen Aufwand an Ausrüstung und Technologie zu gelangen. Keines der ursprünglichen OTA-Gesetze erwähnte etwas anderes als „Plattformen, Systeme, Komponenten oder Materialien“, die „die Einsatzeffektivität des Militärpersonals verbessern“ sollen.
Doch fünf Jahre vor Covid wurde eine außergewöhnliche Nutzung von OTA eingeführt:
In 2015, DoD angekündigt die Gründung des CBRN Medical Countermeasure Consortium, dessen Zweck darin bestand, den OTA-Akquisitionsweg zu nutzen, um „mit dem Verteidigungsministerium zusammenzuarbeiten, um von der FDA lizenzierte chemische, biologische, radiologische und nuklearmedizinische Gegenmaßnahmen zu entwickeln“. [FDA = Food & Drug Administration]
Wie in der Ankündigung von 2015 beschrieben, umfasste dies „Prototyptechnologien für therapeutische medizinische Gegenmaßnahmen, die auf virale, bakterielle und biologische Toxinziele abzielen, die für das Verteidigungsministerium von Interesse sind“. Die Liste der Erreger umfasste die häufigsten Biokriegserreger wie Anthrax, Ebola und Marburg.
In der Ankündigung heißt es weiter: „Zu den Grundlagentechnologien können Tiermodelle viraler, bakterieller oder biologischer Toxinerkrankungen und Pathogenese (mehrere Expositionswege), Assays, Diagnosetechnologien oder andere Plattformtechnologien gehören, die für die Entwicklung zugelassener oder lizenzierter MCMs eingesetzt werden können.“ [medizinische Gegenmaßnahmen].“
Obwohl dies immer noch nicht nach der Produktion von 100 Millionen neuartigen Impfstoffen für den zivilen Gebrauch klingt, bietet es der OTA mehr Spielraum als die sehr begrenzte andere Transaktionsbefugnis des HHS.
Während das HHS OTA die Einhaltung umfassender Entwicklungs- und Herstellungsvorschriften erfordert, erfordert der OTA-Weg für das DoD zur Entwicklung medizinischer Gegenmaßnahmen lediglich eine „FDA-Lizenz“.
Somit wäre es theoretisch möglich, mithilfe der DoD Other Transaction Authorities jegliche Sicherheitsvorschriften zu umgehen – abhängig von den Anforderungen für die FDA-Lizenzierung eines OTA-generierten Produkts. Wie wir sehen werden, wurde im Fall der Covid-mRNA-Impfstoffe eine Notfallgenehmigung erteilt, die keinerlei rechtliche Sicherheitsaufsicht erfordert.
Notfallgenehmigung (EUA)
Hier erfahren Sie, wie die Food & Drug Administration vorgeht (FDA) beschreibt seine EUA-Befugnisse:
Abschnitt 564 des FD&C Act (21 USC 360bbb–3) ermöglicht es der FDA, den Schutz der öffentlichen Gesundheit vor biologischen, chemischen, nuklearen und radiologischen Stoffen zu stärken.
Mit dieser EUA-Berechtigung kann die FDA dazu beitragen, sicherzustellen, dass in Notfällen medizinische Gegenmaßnahmen zur Diagnose, Behandlung oder Vorbeugung schwerer oder lebensbedrohlicher Krankheiten oder Zustände eingesetzt werden können, die durch biologische, chemische, nukleare oder radiologische Stoffe verursacht werden, wenn keine geeigneten, zugelassenen Maßnahmen vorliegen und verfügbare Alternativen (neben anderen Kriterien).
Es ist äußerst wichtig zu verstehen, dass diese EUA-Befugnisse im Jahr 2004 unter sehr spezifischen Umständen im Zusammenhang mit der Vorbereitung auf Angriffe mit Massenvernichtungswaffen, auch bekannt als CBRN-Kampfstoffe (chemische, biologische, radiologische, nukleare), gewährt wurden.
Wie erklärt im Bill of Health von Harvard Law,
Letztlich war es der Krieg gegen den Terror, der zu einer Notfallgenehmigung führte. Nach den Ereignissen vom 11. September 2001 und den darauffolgenden Anthrax-Postanschlägen erließ der Kongress das Project Bioshield Act von 2004. Das Gesetz forderte Milliarden von Dollar an Mitteln für den Kauf von Impfstoffen zur Vorbereitung eines Bioterroranschlags und für die Bevorratung von Notfall-Gegenmaßnahmen. Um im Notfall schnell handeln zu können, erlaubte der Kongress der FDA, formell nicht zugelassene Produkte für den Notfalleinsatz gegen eine Bedrohung der öffentlichen Gesundheit und Sicherheit zuzulassen (vorbehaltlich einer Notstandserklärung des HHS). Der Rekord weist darauf hin, dass sich der Kongress speziell auf die Bedrohung durch Bioterror konzentrierte und nicht auf die Vorbereitung auf eine natürlich auftretende Pandemie.
Das Wortlaut des EUA-Gesetzes unterstreicht die Tatsache, dass es für den Einsatz in Situationen mit Massenvernichtungswaffen gedacht war. Hier sind die 4 Situationen, in denen EUA ausgestellt werden kann:
- eine Feststellung des Heimatschutzministers, dass ein innerstaatlicher Notfall vorliegt oder ein erhebliches Potenzial für einen innerstaatlichen Notfall besteht, der ein erhöhtes Risiko eines Angriffs mit einem oder mehreren biologischen, chemischen, radiologischen oder nuklearen Kampfstoffen birgt;
- eine Feststellung des Verteidigungsministers, dass ein militärischer Notfall vorliegt oder ein erhebliches Potenzial für einen militärischen Notfall besteht, der ein erhöhtes Risiko für United mit sich bringt Staaten Streitkräfte, einschließlich Personal, das unter der Autorität von Titel 10 oder Titel 50 operiert, eines Angriffs mit –
- ein oder mehrere biologische, chemische, radiologische oder nukleare Stoffe; oder
- ein oder mehrere Vertreter, die ein unmittelbar lebensbedrohliches und spezifisches Risiko für United darstellen können oder anderweitig damit verbunden sind Staaten Streitkräfte;
- eine Bestimmung durch die Sekretärin dass ein Notfall im Bereich der öffentlichen Gesundheit vorliegt oder ein erhebliches Potenzial für einen Notfall im Bereich der öffentlichen Gesundheit besteht, der die nationale Sicherheit oder die Gesundheit und Sicherheit von United beeinträchtigt oder erheblich beeinträchtigen kann Staaten im Ausland lebende Bürger, bei denen es sich um einen oder mehrere biologische, chemische, radiologische oder nukleare Wirkstoffe oder eine Krankheit oder einen Zustand handelt, der auf diesen oder diese Wirkstoffe zurückzuführen sein könnte; oder
- die Feststellung einer materiellen Bedrohung gemäß Abschnitt 319F–2 des Gesetz über das öffentliche Gesundheitswesen [42 USC 247d–6b] ausreichend, um die nationale Sicherheit oder die Gesundheit und Sicherheit der Vereinigten Staaten zu beeinträchtigen Staaten im Ausland lebende Bürger.
Nirgendwo in diesen vier Situationen wird eine natürlich auftretende Epidemie, Pandemie oder eine andere Art von Gesundheitsproblem erwähnt, die nicht durch „biologische, chemische, radiologische oder nukleare Stoffe“ verursacht wird.
Könnte SARS-CoV-2 als ein solcher Erreger gelten?
Wenn Sie nach der Definition von „Biologische MittelIm US-Gesetzbuch gehen Sie den folgenden Weg:
Verbrechen und Strafverfahren -> Verbrechen -> Biologische Waffen -> Definitionen
Im Kontext des US-amerikanischen Rechts bedeutet der Begriff „biologische Arbeitsstoffe“ biologische Waffen, und der Einsatz solcher Arbeitsstoffe/Waffen wird als Straftat angesehen.
Wikipedia bietet dies Definition:
Ein biologischer Wirkstoff (auch Bioagent, biologischer Bedrohungsstoff, biologischer Kampfstoff, biologische Waffe oder Biowaffe genannt) ist ein Bakterium, Virus, Protozoon, Parasit, Pilz, oder Toxin, das gezielt als Waffe eingesetzt werden kann Bioterrorismus or biologische Kriegsführung (SW).
Auf welcher Rechtsgrundlage wurde die EUA für Covid-mRNA-Impfstoffe ausgestellt?
Basierend auf den EUA-Gesetzen scheint es, dass keine der vier im Gesetz beschriebenen möglichen Situationen auf ein Produkt angewendet werden könnte, das der Vorbeugung oder Behandlung einer durch einen natürlich vorkommenden Krankheitserreger verursachten Krankheit dienen soll.
Dennoch wurde dieses Gesetz zur Zulassung der mRNA-Covid-Impfstoffe genutzt.
Angesichts der vier im EUA-Gesetz aufgeführten Möglichkeiten war dies diejenige, die für Covid-„Gegenmaßnahmen“ genutzt wurde
C) eine Feststellung durch die Sekretärin dass ein Notfall im Bereich der öffentlichen Gesundheit vorliegt oder ein erhebliches Potenzial für einen Notfall im Bereich der öffentlichen Gesundheit besteht, der die nationale Sicherheit oder die Gesundheit und Sicherheit von United beeinträchtigt oder erheblich beeinträchtigen kann Staaten Im Ausland lebende Bürger, bei denen es sich um einen biologischen, chemischen, radiologischen oder nuklearen Wirkstoff oder um eine Krankheit oder einen Zustand handelt, der auf diesen oder diese Wirkstoffe zurückzuführen sein könnte.
Wann speziell auf Covid angewendet, so wurde es formuliert:
Der Minister des Ministeriums für Gesundheit und menschliche Dienste (HHS) stellte fest, dass ein Notfall im Bereich der öffentlichen Gesundheit vorliegt, der ein erhebliches Potenzial hat, die nationale Sicherheit oder die Gesundheit und Sicherheit von im Ausland lebenden US-Bürgern zu beeinträchtigen, und bei dem es sich um das Virus handelt, das das Coronavirus verursacht Krankheit 2019 (COVID-19)…
Hier besteht kein Zweifel daran, dass „das Virus, das COVID-19 verursacht“ als Äquivalent zu „einem oder mehreren biologischen, chemischen, radiologischen oder nuklearen Wirkstoffen“ gilt.
Es ist auch wichtig zu beachten, dass die „Feststellung eines Notfalls im Bereich der öffentlichen Gesundheit“ durch die EUA völlig unabhängig von anderen Notfallerklärungen im Bereich der öffentlichen Gesundheit ist und in keiner Weise von diesen abhängt, wie etwa denen, die von der WHO oder der US-Regierung abgegeben wurden , und der Präsident zu Beginn der Covid-19-Pandemie.
Selbst wenn also die WHO, die US-Regierung und der Präsident erklären, dass die Pandemie vorbei ist, kann es immer noch eine Notfallgenehmigung geben, wenn der HHS-Sekretär weiterhin behauptet, dass die in Abschnitt C) beschriebene Situation vorliegt.
Mit Blick auf alle EUAs für Hunderte von Covid-bezogenen Medizinprodukten, ist es sehr schwer zu erkennen, wie der HHS-Sekretär die Behauptung rechtfertigen könnte, dass „es einen Notfall im Bereich der öffentlichen Gesundheit gibt, der ein erhebliches Potenzial hat, die nationale Sicherheit oder die Gesundheit und Sicherheit von im Ausland lebenden US-Bürgern zu beeinträchtigen“ in den meisten, wenn nicht allen, dieser Fälle.
Zusätzliche „gesetzliche Kriterien“ für die Erteilung einer Notfallgenehmigung durch die FDA
Sobald der HHS-Sekretär erklärt, dass ein Notfall im Bereich der öffentlichen Gesundheit vorliegt, der eine EUA erfordert, basierend auf einer der vier im Gesetz aufgeführten Situationen, müssen vier weitere „gesetzliche Kriterien“ erfüllt sein, damit die FDA die EUA ausstellen kann . So erklärt die FDA diese Anforderungen:
- Schwere oder lebensbedrohliche Krankheit oder Zustand
Damit die FDA eine EUA ausstellen kann, müssen die in der EUA-Erklärung des HHS-Sekretärs genannten CBRN-Wirkstoffe in der Lage sein, eine schwere oder lebensbedrohliche Krankheit oder einen schweren oder lebensbedrohlichen Zustand zu verursachen.
HINWEIS: Dieses Kriterium wiederholt die Spezifikation eines CBRN-Wirkstoffs, der gesetzlich als Waffe definiert ist, die zur Begehung einer Straftat verwendet wird.
- Wirksamkeitsnachweis
Medizinische Produkte, die für eine EUA in Betracht gezogen werden können, sind solche, die „wirksam sein können“, um schwere oder lebensbedrohliche Krankheiten oder Zustände zu verhindern, zu diagnostizieren oder zu behandeln, die durch einen oder mehrere CBRN-Wirkstoffe verursacht werden können, die in der Erklärung des HHS-Sekretärs aufgeführt sind Notfall oder drohender Notfall gemäß Abschnitt 564(b).
Der „kann wirksam sein“-Standard für EUAs sieht ein geringeres Maß an Beweisen vor als der „Wirksamkeits“-Standard, den die FDA für Produktzulassungen verwendet. Die FDA beabsichtigt, die potenzielle Wirksamkeit eines möglichen EUA-Produkts von Fall zu Fall anhand einer Risiko-Nutzen-Analyse zu bewerten, wie unten erläutert.
[Fettschrift hinzugefügt]
RECHTLICHE FRAGE: Wie kann jemand rechtlich behaupten, dass ein gemäß EUA zugelassenes Produkt „sicher und wirksam“ ist, wenn der gesetzliche Standard für EUA „könnte wirksam sein“ lautet und die FDA erklärt, dass dies ein „geringerer Beweisgrad“ als der verwendete Standard ist? für regelmäßige Produktzulassungen?
- Risiko-Nutzen-Analyse
Ein Produkt kann für eine EUA in Betracht gezogen werden, wenn der Kommissar feststellt, dass die bekannten und potenziellen Vorteile des Produkts bei Verwendung zur Diagnose, Vorbeugung oder Behandlung der identifizierten Krankheit oder des festgestellten Zustands die bekannten und potenziellen Risiken des Produkts überwiegen.
Bei der Feststellung, ob die bekannten und potenziellen Vorteile des Produkts die bekannten und potenziellen Risiken überwiegen, hat die FDA will schauen die Gesamtheit der wissenschaftlichen Erkenntnisse heranzuziehen, um eine Gesamtrisiko-Nutzen-Beurteilung vorzunehmen. Solche Beweise, die entstehen könnte aus verschiedenen Quellen, könnte beinhalten (ist aber nicht beschränkt auf): Ergebnisse in- und ausländischer klinischer Studien, In-vivo-Wirksamkeitsdaten aus Tiermodellen und In-vitro-Daten, zur Prüfung durch die FDA verfügbar. Die FDA wird auch die Qualität und Quantität bewerten verfügbare Beweise, angesichts des aktuellen Stands der wissenschaftlichen Erkenntnisse.
[Fettschrift hinzugefügt]
RECHTLICHER HINWEIS: Es gibt keine gesetzliche Norm und es gibt keine rechtlichen Definitionen dafür, was es bedeutet, dass „bekannte und potenzielle Vorteile“ die „bekannten und potenziellen Risiken“ überwiegen. Es gibt auch keine qualitative oder quantitative rechtliche Definition dafür, was akzeptable „verfügbare Beweise“ sind, auf denen die Risiko-Nutzen-Analyse „basieren“ kann. Es könnte keine tatsächlichen Beweise geben, aber die Überzeugung, dass ein Produkt einen großen potenziellen Nutzen und kein großes potenzielles Risiko birgt, würde diese „gesetzliche Anforderung“ erfüllen.
- Keine Alternativen
Damit die FDA eine EUA ausstellen kann, darf es keine angemessene, zugelassene und verfügbare Alternative zum Kandidatenprodukt zur Diagnose, Vorbeugung oder Behandlung der Krankheit oder des Leidens geben. Ein potenzielles Alternativprodukt kann als „nicht verfügbar“ betrachtet werden, wenn nicht genügend Vorräte der zugelassenen Alternative vorhanden sind, um den Notfallbedarf vollständig zu decken.
RECHTLICHE FRAGE: Abgesehen von der ungeheuerlichen und möglicherweise kriminellen Verunglimpfung/Verbot alternativer Covid-19-Behandlungen wie Ivermectin und Hydroxychloroquin: Zu welchem Zeitpunkt gab es eine zugelassene Alternative zur „Prävention von Covid-19“ (die einzige Aufgabe, zu der die mRNA-Impfstoffe gekauft wurden)? ) – zum Beispiel Paxlovid – was dazu führen würde, dass eine EUA für die mRNA-Impfstoffe nicht mehr legal ist?
Hier erfahren Sie, wie alle diese „gesetzlichen Kriterien“ tatsächlich erfüllt wurden Notfallgenehmigung für die Covid-mRNA-Impfstoffe von BioNTEch/Pfizer:
Ich bin zu dem Schluss gekommen, dass die Notfallverwendung des Pfizer-BioNTech COVID-19-Impfstoffs zur Vorbeugung von COVID-19 bei Verabreichung wie im Zulassungsumfang (Abschnitt II) beschrieben die Kriterien für die Erteilung einer Zulassung gemäß Abschnitt 564(c) erfüllt das Gesetz, weil:
- SARS-CoV-2 kann bei Menschen, die mit diesem Virus infiziert sind, eine schwere oder lebensbedrohliche Krankheit oder Erkrankung, einschließlich schwerer Atemwegserkrankungen, verursachen;
- Basierend auf der Gesamtheit der der FDA zur Verfügung stehenden wissenschaftlichen Beweise ist es berechtigt anzunehmen, dass der Pfizer-BioNTech COVID-19-Impfstoff kann bei der Vorbeugung von COVID-19 wirksam seinund dass bei Verwendung unter den in dieser Zulassung beschriebenen Bedingungen die bekannten und potenziellen Vorteile des Pfizer-BioNTech COVID-19-Impfstoffs bestehen bei Verwendung zur Vorbeugung von COVID-19 die bekannten und potenziellen Risiken überwiegen; Und
- Es gibt keine adäquate, zugelassene und verfügbare Alternative zum Notfalleinsatz des Pfizer-BioNTech COVID-19-Impfstoffs um COVID-19 zu verhindern.
[Fettschrift hinzugefügt]
HINWEIS: Der einzige Kontext, in dem die FDA die potenziellen Vorteile und Risiken des Impfstoffs abwog und in dem sie feststellte, dass er „wirksam sein könnte“, war bei der Prävention von Covid-19.
Es gibt keine Überlegungen, keine Belege für einen tatsächlichen oder potenziellen Nutzen und keine Feststellung, ob der Impfstoff eine potenzielle Wirksamkeit für irgendetwas anderes hat, einschließlich: Senkung des Risikos einer schweren Erkrankung, Senkung des Risikos einer Krankenhauseinweisung, Senkung des Sterberisikos , wodurch das Risiko von Erkrankungen verringert wird, die tatsächlich oder potenziell mit Covid-19 in Zusammenhang stehen.
Daher könnte man berechtigterweise die Rechtmäßigkeit aller Behauptungen in Frage stellen, dass der Impfstoff „sicher und wirksam“ sei, und zwar im Zusammenhang mit etwas anderem als „bei Verwendung zur Vorbeugung von COVID-19“ – wovon die Impfstoffe sehr bald, nachdem sie bekannt waren, NICHT taugen eingeführt.
Wenn man den Menschen sagen würde, dass die mRNA-Impfstoffe von BioNTech/Pfizer zu etwas anderem als der Vorbeugung von Covid-19 „sicher und wirksam“ seien, und wenn ihnen irgendwelche Konsequenzen für die Nichteinnahme des Impfstoffs zu einem anderen Zweck als der Vorbeugung von Covid-19 angedroht würden, dann könnten sie das tun Haben Sie ein berechtigtes Argument dafür, dass sie aufgrund betrügerischer Behauptungen illegal zur Einnahme eines nicht zugelassenen Produkts gezwungen wurden?
Anforderungen der dritten Stufe für EUA für nicht zugelassene Produkte
Sobald wir die EUA-spezifische Notfallerklärung haben und sobald die FDA erklärt, dass das Produkt möglicherweise wirksam ist und dass alle verfügbaren Beweise (von null bis unendlich) zeigen, dass sein Nutzen seine Risiken überwiegt (wie durch die Einschätzung der FDA bestimmt). sein), gibt es eine weitere Ebene nicht sicherheits- und nichtwirksamkeitsbezogener Vorschriften.
Hier ist, wie ein Bericht des Congressional Research Service 2018 über EUA erklärt das:
FFDCA §564 weist die FDA an, bestimmte erforderliche Bedingungen in einer EUA aufzuerlegen, und lässt gegebenenfalls zusätzliche Ermessensbedingungen zu. Die erforderlichen Bedingungen variieren je nachdem, ob es sich bei der EUA um ein nicht zugelassenes Produkt oder um die nicht genehmigte Verwendung eines zugelassenen Produkts handelt. Für ein nicht zugelassenes Produkt müssen die Nutzungsbedingungen:
(1) sicherstellen, dass medizinische Fachkräfte, die das Produkt verabreichen, die erforderlichen Informationen erhalten;
(2) sicherstellen, dass Personen, denen das Produkt verabreicht wird, die erforderlichen Informationen erhalten;
(3) für die Überwachung und Meldung unerwünschter Ereignisse im Zusammenhang mit dem Produkt sorgen; Und
(4) die Führung von Aufzeichnungen und die Berichterstattung durch den Hersteller vorsehen.
RECHTLICHE FRAGE: Was genau sind die „erforderlichen Informationen“? Wir wissen, dass die Menschen darüber informiert wurden, dass den Impfstoffen eine Notfallgenehmigung erteilt wurde. Aber wurde ihnen gesagt, dass dies „ein geringeres Maß an Beweisen“ bedeute, als für „sichere und wirksame“ Aussagen zu anderen Medizinprodukten erforderlich sei? Wurden sie darüber informiert, dass es unterschiedliche Stufen von „sicher und wirksam“ gibt, je nachdem, ob ein Produkt über eine EUA- oder eine andere Art von Zulassung verfügt?
HINWEIS: Das Gesetz verlangt, dass es eine Möglichkeit gibt, unerwünschte Ereignisse zu überwachen und zu melden. Es wird jedoch nicht angegeben, wer überwacht, welche Standards für die Berichterstattung gelten und wie hoch die Schwelle für die Ergreifung von Maßnahmen auf der Grundlage der Berichte ist.
EUA im Vergleich zu allen anderen Zulassungswegen für Arzneimittel/Impfstoffe
Als Forscher/Autor Sascha Latypova hat darauf hingewiesen, dass viele Menschen von EUA verwirrt waren, weil es sehr nach EAU klingt, was für „Expanded Access Use“ steht. Hierbei handelt es sich um eine Art der Zulassung von Medizinprodukten, wenn ein dringender Bedarf einer bestimmten Gruppe von Patienten besteht (z. B. Krebspatienten im Stadium IV, deren Lebenserwartung in Monaten gemessen wird), die bereit sind, im Gegenzug für den Zugang unerwünschte Ereignisse und sogar den Tod zu riskieren zu einer experimentellen Behandlung.
Die Genehmigung zur Notfallnutzung steht in keinem Zusammenhang mit der Nutzung des erweiterten Zugangs und hat auch keine Ähnlichkeit damit.
Die verschiedenen rechtlichen Wege zur Zulassung von Medizinprodukten werden in einer von Rechtsforschern hervorgehobenen Tabelle übersichtlich dargestellt Katharina Watt. Die Tabelle ist Teil einer Präsentation im Jahr 2020 für eine gemeinsame Lernsitzung von FDA und CDC: Regulatorische Aktualisierungen zum Einsatz medizinischer Gegenmaßnahmen.
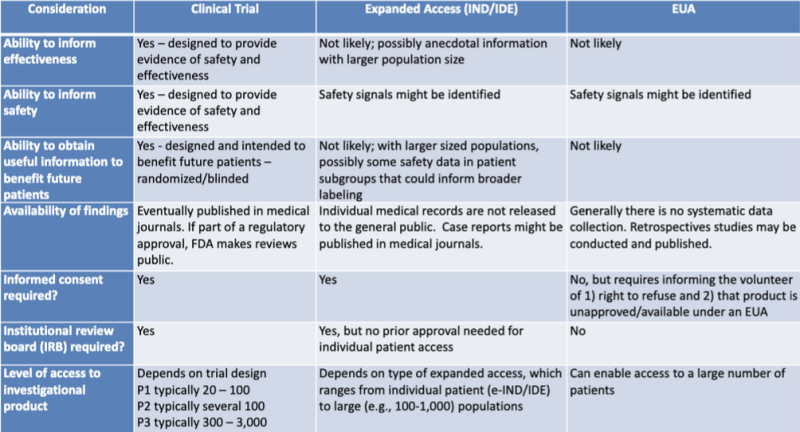
Diese Tabelle zeigt sehr deutlich, dass der EUA-Prozess wahrscheinlich keine Informationen über die Produktwirksamkeit liefern wird, nicht darauf ausgelegt ist, Sicherheitsnachweise zu liefern, wahrscheinlich keine nützlichen Informationen zum Nutzen zukünftiger Patienten liefern wird, keine systematische Datenerfassung beinhaltet, keine retrospektiven Studien erfordert, keine Einverständniserklärung und kein institutioneller Prüfungsausschuss.
Darüber hinaus in a 2009 Institute of Medicine of the National Academic Veröffentlichung, ebenfalls hervorgehoben von Watt, mit dem Titel „Medizinische Gegenmaßnahmen: Dispensing Emergency Use Authorization and the Postal Model – Workshop Summary“ finden wir diese Aussage auf S. 28:
Es ist wichtig zu erkennen, dass eine EUA nicht Teil des Entwicklungspfades ist; Dabei handelt es sich um eine völlig separate Einheit, die nur in Notfällen zum Einsatz kommt und nicht Teil des Arzneimittelzulassungsverfahrens ist.
Bedeutet das, dass Zulassungen von Covid-19-Gegenmaßnahmen, die auf EUAs basierten, rechtswidrig waren? Bedeutet das, dass es keine rechtliche Möglichkeit gibt, zu behaupten, ein EUA-Produkt sei „sicher und wirksam“, weil es NICHT TEIL des ARZNEIMITTELZULASSUNGSPROZESSES ist?
Zusammenfassung
Angesichts aller Informationen in diesem und im vorangehenden Artikel ist dies deutlich zu erkennen Teil 1, dass die Covid-mRNA-Impfstoffe von BioNTach/Pfizer nach Militärgesetzen entwickelt, hergestellt und zugelassen wurden, die für Notsituationen im Zusammenhang mit biologischer Kriegsführung/Terrorismus und nicht für natürlich vorkommende Krankheiten, die die gesamte Zivilbevölkerung betreffen, vorbehalten sind.
Daher war die Einhaltung von Vorschriften und die Aufsicht, die wir erwarten, wenn ein Produkt für die gesamte Zivilbevölkerung als „sicher und wirksam“ gilt, gesetzlich nicht vorgeschrieben.
Kann diese Analyse verwendet werden, um die Rechtmäßigkeit der Behauptung „sicher und wirksam“ jener Regierungsbeamten in Frage zu stellen, die wussten, was EUA beinhaltete? Gibt es weitere rechtliche Konsequenzen?
Hoffentlich.
Wichtig ist, dass es bei den rechtlichen Anfechtungen von Covid-mRNA-Impfstoffen bisher (soweit mir bekannt ist) keine Entscheidungen darüber gibt, ob Militärrecht wie OTA und EUA auf zivile Situationen angewendet werden kann. Es gab jedoch eine Erklärung des Bezirksrichters Michael Truncale in seinem Abweisung des Falls des Whistleblowers Brook Jackson gegen Ventavia und Pfizer, das ist wichtig zu bedenken.
Hier erkennt der Richter an, dass es sich bei der Vereinbarung für die mRNA-Impfstoffe von BioNTech/Pfizer um ein militärisches OTA handelte, weigert sich jedoch, über deren Anwendbarkeit auf die nichtmilitärischen Umstände (natürlich vorkommende Krankheit, 100 Millionen Dosen, größtenteils nicht für militärische Zwecke) zu entscheiden, unter denen sie gilt wurde ausgestellt:
Die Tatsache, dass sowohl Militärangehörige als auch Zivilisten den Impfstoff erhielten, bedeutet nicht, dass der Erwerb des Impfstoffs für die Verbesserung der Missionseffektivität des Militärs irrelevant war. Noch wichtiger ist, dass Frau Jackson dieses Gericht faktisch darum bittet, die Entscheidung des Verteidigungsministeriums aufzuheben, andere Transaktionsbefugnisse auszuüben, um den Impfstoff von Pfizer zu kaufen. Aber wie der Oberste Gerichtshof der Vereinigten Staaten seit langem betont, handelt es sich bei den „komplexen subtilen und professionellen Entscheidungen über die Zusammensetzung, Ausbildung, Ausrüstung und Kontrolle einer Streitmacht im Wesentlichen um professionelle militärische Urteile.“ Gilligan gegen Morgan, 413 U.S. 1, 10 (1973). Daher sei es „schwer, sich einen Bereich staatlicher Tätigkeit vorzustellen, in dem die Gerichte über weniger Kompetenz verfügen“. Ausweis. Dieses Gericht wird kein Veto gegen die Urteile des Verteidigungsministeriums zur Wirksamkeit der Mission während eines nationalen Notfalls einlegen.
Dies ist nur eine von vielen rechtlichen Hürden, die im Kampf um ein endgültiges Verbot aller während des Covid-19-Notstands zugelassenen mRNA-Produkte und aller nachfolgenden mRNA-Produkte, deren Zulassung auf dem Covid-19-Zulassungsverfahren basierte, bestehen bleiben.
Veröffentlicht unter a Creative Commons Namensnennung 4.0 Internationale Lizenz
Für Nachdrucke setzen Sie bitte den kanonischen Link wieder auf das Original zurück Brownstone-Institut Artikel und Autor.








