

In den letzten Jahrzehnten meiner Karriere habe ich unzählige Stunden damit verbracht, mich für den Schutz der Amerikaner einzusetzen, indem ich die Sicherheit von Arzneimitteln erforschte. Meine Ausbildung und Karriere führten mich zu etwa einem halben Dutzend Universitäten, zu Big Pharma und zur FDA unter drei Präsidentenverwaltungen. Bei der Arzneimittelsicherheit wird berücksichtigt, warum eine Person möglicherweise ein Arzneimittel einnimmt und keine Nebenwirkungen hat, während eine andere Person das gleiche Produkt einnehmen könnte, aber Nebenwirkungen bis hin zur dauerhaften Behinderung oder zum Tod hat. Bei der Untersuchung der Arzneimittelsicherheit werden standardmäßig auch nichtklinische Aspekte der Herstellung und Arzneimittelqualität berücksichtigt.
Da die Arzneimittelqualität ein wesentlicher Faktor bei der Beurteilung der Arzneimittelsicherheit ist, führte mein Einsatz zum Schutz der Amerikaner dazu, dass ich das weltweit erste Unternehmen konzipierte und gründete.analytische Apotheke„hat die Aufgabe, pharmazeutische Produkte aus Ländern wie Indien und China wissenschaftlich zu überprüfen, bevor sie an Patienten abgegeben werden. Leider führte das Streben nach Freizügigkeit gegenüber Ethik und Patientenschutz dazu, dass sich das Finanzmanagement dieses Unternehmens verpflichtete umfangreiche Verstöße gegen die FDA und von Richtern beschuldigt zu werden falsche wissenschaftliche Behauptungen (was alles zufällig nach meinem Ausstieg geschah).
Ohne externe Bestätigung der Arzneimittelqualität sind die Amerikaner bei der Beurteilung und Bestätigung der Produktreinheit vollständig auf die FDA und die Hersteller angewiesen. Es hat sich gezeigt, dass die Arzneimittelsicherheit ein erhebliches Problem darstellt, wenn es um Covid-mRNA-Injektionen geht. Wenn jemand seine eigene Analyse zu mRNA-Injektionen durchführen wollte, dann leider Sie verfügen nicht über eine entsprechend detaillierte Zutatenliste, mit der Sie sie vergleichen könnten, oder haben nicht einmal Zugriff auf die etablierte Regulierungsmethodik, wie sie ordnungsgemäß auf Reinheit getestet werden kann.
Es liegt an den Herstellern und Die FDA berücksichtigt alle Bestandteile dieser mRNA-Injektionen, einschließlich der Reihenfolge der Eigenschaften von mRNA und Lipidnanopartikeln (LNP), einschließlich Halbwertszeit, LNP-Strukturen, Oberflächenmodifikation(en), Anzahl/Art(en) von LNPs pro Dosis und Befestigungspunkten der mRNA-Strang, nicht spezifiziert oder „Geschäftsgeheimnis“.
Darüber hinaus berücksichtigt die FDA zusätzlich die Methodologien Auch die Frage, wie man mRNA-Injektionen auf Reinheit prüft, ist ein Geschäftsgeheimnis.
Überparteiliche Unterstützung und Hunderte Milliarden Steuergelder, aber KEINE Transparenz?
Die Covid-mRNA-Geheimhaltung besteht, obwohl sowohl die Trump- als auch die Biden-Regierung vollständige Transparenz bei mRNA-Injektionen bis hin zur Aufhebung der geistigen Eigentumsrechte an Covid-mRNA vorgeschlagen hatten. Dennoch erlauben sowohl die FDA als auch die Hersteller Patente, einschließlich grundlegender Daten zu diesen Impfungen, als Geschäftsgeheimnis bzw. halten sie streng im Griff. Sie tun dies, obwohl alle Hersteller von Covid-Impfstoffen diesen erhalten haben Hunderte Millionen Steuergelder nach Forbes/Statista Veröffentlichungen.
Das Studium der Arzneimittelsicherheitsepidemiologie ist schon schwierig genug. Ohne nachweisbare Produktreinheit/-konsistenz ist eine vollständige Sicherheitsbewertung nicht möglich.
Vollständige Transparenz aller Inhaltsstoffe und Maßnahmen zur Qualitätskontrolle sind nicht nur deshalb wichtig, weil sie mit Hunderten Millionen Dollar stark aus Steuergeldern finanziert wurden, sondern auch, weil eine Reihe von Fragen zur Sicherheit und Wirksamkeit von Covid-mRNA-Injektionen aufgekommen sind.
Ihre Genehmigung war nicht nur außerordentlich komplex, sondern wurde auch von den Aufsichtsbehörden beschleunigt weniger als ein Jahr. Die meisten Medikamente und Impfstoffe dauern normalerweise ca 10 Jahre um die Sicherheit/Wirksamkeit vollständig zu testen und zu überprüfen und zu genehmigen. Darüber hinaus sind die Inhaltsstoffe völlig neu, sehr komplex und die ersten ihrer Art, die in großem Umfang verabreicht werden, einschließlich der Entwicklung Langzeitbewertungen der klinischen Sicherheit/Toxizität und epidemiologische Überprüfungen wurden beschleunigt und waren vor der Veröffentlichung wahrscheinlich nicht vollständig geklärt.
Die Überprüfung von Inhaltsstoffen, Transparenz und „Wahrhaftigkeit“ der FDA gibt es schon seit dem 1800. Jahrhundert:
Die analytische Überprüfung und die Transparenz der Inhaltsstoffe oder „Wahrheit bei der Etikettierung“, wo sich der Inhalt der Flasche befindet falls angefordert passend zu den aufgeführten Zutaten stammt aus der Zeit vor der Gründung der FDA im Jahr 1862. Die heutige FDA entstand eigentlich aus einem einzigen Mitarbeiter des „Department of Chemistry“, der im US-Landwirtschaftsministerium beschäftigt war.
Verfälschung, (veränderte oder giftige Inhaltsstoffe) falsches Branding (ein falsches Etikett enthält oder anderweitig irreführend ist oder falsche medizinische Angaben enthält) oder Fehlbezeichnung (enthält Inhaltsstoffe, die nicht auf dem Produktetikett aufgeführt sind) haben alle eine lange, hässliche Geschichte in Amerika. Es wurde angenommen, dass die Ungeheuerlichkeit Anfang bis Mitte des 19. Jahrhunderts ihren Höhepunkt erreicht hatte – oder zumindest zu diesem Zeitpunkt erkennbar wurde –, da erst 1862 technische Verfahren zur Analyse und Erkennung von Zutatenbetrug entwickelt wurden. Zuvor verkauften sogenannte „reisende Medizinmänner“, die sich selbst „Ärzte“ nannten (ausnahmslos mit zweifelhaften oder nicht vorhandenen Qualifikationen), Flaschen mit „Allheilmittel“-Produkten, deren Inhaltsstoffetiketten nur nebulöse oder harmlose Inhalte aufführten, wie z „Vitamine""Kräuterextrakte," oder "Schlangenöl“ – oder haben oft überhaupt keine Zutatenliste.
Damals gab es viele gläubige, puritanische Neu-Engländer, die das aus religiösen Gründen tun würden hört niemals Wer mit Alkohol in Berührung kam, kaufte diese Lösungen von diesen Händlern und ließ sich unwissentlich dazu verleiten, Lösungen zu konsumieren, die nicht nur Alkohol, sondern auch Betäubungsmittel wie Opium und/oder Kokain enthielten. Unter dem Vorwand, ein unglaublich breites Spektrum an Beschwerden zu lindern, entwickelten die Patienten stattdessen eine bestrafende Sucht und/oder ihre Gesundheit wurde durch diese frühen „Drogendealer“ auf andere Weise negativ beeinflusst.
Als das Problem zunahm, wurde die Bundesregierung darauf aufmerksam. Schließlich ist die Pure Food and Drug Act wurde 1906 verabschiedet und führte zur Gründung der Food and Drug Administration (FDA).
[Die FDA hatte eine Ausbildung Die Pflicht besteht darin, sicherzustellen, dass Arzneimittel wahrheitsgemäße Angaben auf dem Etikett enthalten und bestimmte Reinheits- und Stärkestandards erfüllen.
Denken Sie daran, dass fast 120 Jahre alt wahrheitsgetreue Kennzeichnungspflicht und „Reinheit“-Teil des Pure Food and Drug Act von 1906, während Sie weiter über mRNA-Verifizierungstests und Inhaltsstoffetransparenz lesen.]
Welche „wahrheitsgemäßen“ und „reinen“ Prüfungen zur Inhaltsstoffverifizierung werden für von der FDA regulierte Produkte durchgeführt?
Bereits im Jahr 2021 beschloss die FDA, mit der Überwachung der pharmazeutischen Qualität Amerikas über eine zu beginnen Fernabholung of Einsendung der Proben per Post für Medikamente als Ersatz für Live-Inspektionen in Einrichtungen aufgrund der Covid-Pandemie. War das legal? Könnte das jemals als wissenschaftlich angemessen angesehen werden? Trotz des Endes der Pandemie findet heute die einzige offizielle Arzneimittelfreigabeprüfung statt, die derzeit durchgeführt wird jedem Covid-mRNA-Arzneimittel erscheint zu Noch Dies kann von der FDA über ein vom Hersteller bereitgestelltes Verfahren erfolgen.per Post eingeschickt“ Probe nach a Screenshot der aktuellen FDA-Website. Offensichtlich unterscheidet sich eine per Post zugesandte Probenahmemethode erheblich und ist möglicherweise weniger zuverlässig als die direkte Probenentnahme über eine direkte, persönliche Entnahmemethode. Trotzdem behauptet die FDA, dass sie „der weltweit höchste Standard für Probenahme und Prüfung"
Darüber hinaus schlägt die FDA vor, ihre „Mailed-In“-Ferntestrichtlinie weiter voranzutreiben neu vorgeschlagenes Leitliniendokument.
Obwohl es nur als „Entwurf“ eines FDA-Dokuments existiert, zeigen offizielle FDA-Websites dies Der Probenversand scheint bereits seit mindestens Januar 2021 umgesetzt zu sein. Die FDA scheint die Ergebnisse dieser per Post eingesandten Tests als ihre unabhängige Überprüfung geltend zu machen.
Darüber hinaus unten auf der ersten Seite des FDA-Entwurfs Dokument schlägt Ausweitung von „Ferntests“ vor. Es ist derzeit aufgeführt alles, Dies bedeutet, dass es sich um einen behördenweiten Richtlinienvorschlag handelt.
Die vollständige Liste enthält Folgendes:
- Amt für Regulierungsangelegenheiten
- Büro für Lebensmittelpolitik und -reaktion
- Büro für Kombinationsprodukte
- Zentrum für Bewertung und Forschung von Biologika
- Zentrum für Arzneimittelevaluierung und -forschung
- Zentrum für Geräte und radiologische Gesundheit
- Zentrum für Lebensmittelsicherheit und angewandte Ernährung
- Zentrum für Tabakwaren
- Zentrum für Veterinärmedizin
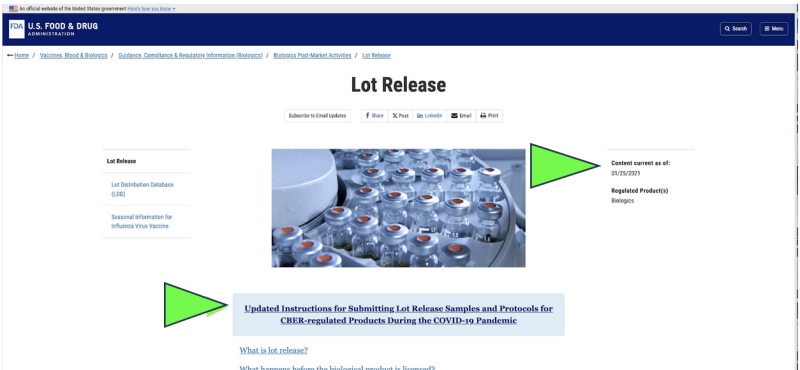
Ist die Qualitätssicherung per Post durch die FDA angemessen? Was wäre, wenn die Restaurantinspektionen des Gesundheitsministeriums der Bundesstaaten die Richtlinien der FDA widerspiegeln würden?
Diese „Mail-in“-Probenahmemethode ist ähnlich absurd, wenn beispielsweise das Gesundheitsamt eines Staates Restaurants überwacht, indem es diese auffordert, regelmäßig verschiedene Artikel aus ihrer Speisekarte an eine Testeinrichtung zu „senden“, damit die Gesundheitsämter Tests auf potenzielle Lebensmittel durchführen können -bedingte Kontamination und/oder die Aufforderung an Restaurants, zu versprechen, Menüpunkte selbst zu testen. Was wäre, wenn dieses Restaurant in China wäre? Was wäre, wenn dieses Restaurant in Indien wäre? Oder jedes andere Land, von dem bekannt ist, dass es eine hat miserable Geschichte von Betrug und Qualitätskontrolle Probleme?
Diese Methodik wäre sowohl für Restaurants als auch für Pharmaunternehmen inakzeptabel, und zwar aus offensichtlichen Gründen: Hersteller könnten die von ihnen bevorzugten Proben einsenden – nicht unbedingt repräsentative Chargenproben. Es ist offensichtlich nicht dasselbe, dass FDA-Inspektoren bei unangekündigten Inspektionen der gesamten Anlage Proben entnehmen.
In der Restaurant-Analogie würden das natürlich alle Restaurants tun Reichen Sie Proben der Note „A“ ein Dies wäre nicht unbedingt repräsentativ für das, was die Verbraucher erhalten.

Qualitätskontrolle: Was ist ein pharmazeutischer „Freisetzungstest“ und warum ist er wichtig?
Heute überwacht die FDA die Qualität und den Inhalt von $2.7 Billionen Produktwert pro Jahr, scheint aber kritische Beurteilungen und Ergebnisse zur Überprüfung von Inhaltsstoffen zu unterdrücken. Die FDA soll durch ihr Verhalten die Amerikaner schützen umfassend analytische Tests als Prüfsumme, um die Genauigkeit der Inhaltsstoffe sicherzustellen. Die Ergebnisse sollten für die Steuerzahler, die das finanzieren, transparent sein 6.6 Milliarden US-Dollar der FDA Budget. Diese wissenschaftliche Überprüfung wird als pharmazeutisch bezeichnet.Release-Tests.“ Release-Test ist ein technischer Begriff, der sich auf einen Prozess bezieht, der eine Vielzahl instrumenteller Analysen umfasst umfassend Testen Sie Produkte auf Reinheit, Konzentration, Konsistenz, Identität und Verunreinigungen jeglicher Art.
Die gesamte FDA entstand aus diesem einen Mitarbeiter der „Abteilung für Chemie“ aus dem Jahr 1862 und dem Bedarf an Transparenz und Überprüfung der Inhaltsstoffe. Heute hat sich dieser Mitarbeiter zu einem entwickelt gesamte FDA-Abteilung mit 1,300 Wissenschaftlern und Hilfspersonal vermutlich der Überprüfung von Inhaltsstoffen durch pharmazeutische Freigabetests gewidmet. Die FDA Büro für pharmazeutische Qualität (OPQ) soll sicherstellen, dass Arzneimittel genau dem Gehalt der aufgeführten Inhaltsstoffe entsprechen, ohne Qualitäts-/Unreinheits- (qualitativ) oder inhaltliche (qualitative) Schwankungen. Die dafür erforderlichen Regeln sind sehr spezifisch und detailliert 21 CFR § 201.10.
Wie die FDA mRNA-Injektionen zur Qualitätskontrolle überprüft:
Die Ergebnisse der Qualitätskontrolle von Tests mit mRNA-Injektionen waren besonders kritisch, da sie umfangreich und komplex sind und schnell durchgeführt wurden. Während Steuerzahler auf die FDA angewiesen sind, um die Qualität der mRNA-Injektion zu überprüfen und die Ergebnisse zu teilen, ist die FDA scheint verpflichtet, die Inhaltsstoffe der Hersteller auf Kosten selbst der grundlegendsten Transparenz in Bezug auf mRNA-Covid-Produkte zu schützen. Während die FDA offenbar Proben sammelt, ist ihre „Mail-In“-Methode grundsätzlich fehlerhaft. Darüber hinaus gibt die FDA die Ergebnisse dieser Tests nirgendwo weiter, wo ich sie finden könnte.
Mit anderen Worten: Während der Pandemie, als brandneue, weit verbreitete mRNA-Impfungen den Amerikanern mit „Warp-Geschwindigkeit“ aufgedrängt wurden und als Amerika sich am meisten auf die Qualitäts-/Regulierungspflichten der FDA verließ, akzeptierte die FDA selbst eingereichte „per Post“ in“ Qualitätskontrolltests und/oder Ergebnisse. Hat die FDA das nicht berücksichtigt? mRNA-Hersteller gaben zu, dass sie „Schwierigkeiten“ hatten, auf die Produktion zu reagieren, und „sich darum bemühten“, mitzuhalten mit Herstellungsprozessen? Hersteller von mRNA-Inhaltsstoffen gaben außerdem an, dass die Bemühungen, den Bedarf zu decken, „beispiellos“ seien.
Aussagen wie diese erwecken kein Vertrauen der Verbraucher in die Qualität und veranschaulichen die enorme Aufwertung dieser komplexen Produkte, die gerechtfertigt sein sollte besonders wachsam und persönliche FDA-Prüfung von Einrichtungen und hergestellten Produkten, ob Pandemie oder nicht. Ein Hersteller von mRNA-Inhaltsstoffen gab beispielsweise an, dass er seine Produktion plötzlich um XNUMX % hochgefahren habe 50 Falte.
Inmitten dieser neuartigen Technologie, die mit „Warp-Geschwindigkeit“ durchgesetzt wurde, forderte keiner der 1,300 OPQ-Wissenschaftler bei der FDA Live-Inspektionen oder bot zumindest an, etwas anderes zu tun, als nach potenziell fragwürdigen „eingeschickten“ Proben zu fragen zum Prüfen?
Die offensichtliche Frage ist: Warum hat die FDA die Proben nicht direkt gesammelt?? Auch wenn die Pandemie im Gange wäre, hätte die FDA Einrichtungen inspizieren können, die Schutzanzüge trugen oder – oder am sehr Zumindest haben sie sich dafür entschieden, Proben in Apotheken, Krankenhäusern oder in den Lagerhäusern der Händler zu sammeln.
Verborgene Methodik zum Testen der Inhaltsstoffe der mRNA-Injektion:
Abgesehen vom Fehlen von Testergebnissen und fragwürdigen „zugesandten“ Probenahmeergebnissen ist dies bei der FDA der Fall zusätzlich Sie verbergen ihre validierte Methodik und hindern andere daran, ihre eigenen, unabhängigen Analysen zur Qualität/Reinheit von mRNA-Injektionen durchzuführen.
Die unabhängige Analyse von Arzneimitteln auf Reinheit und potenzielle Kontamination im Vergleich zur Zutatenliste war etwas, das ich selbst versucht hatte, als ich das weltweit erste Produkt konzipierte analytische Apotheke. Da es sich bei mRNA-Spritzen jedoch um eine neuartige Technologie mit einer nicht völlig transparenten Liste der Inhaltsstoffe handelt, ist die anzuwendende Testmethode nicht so einfach wie bei anderen niedermolekularen Arzneimitteln. Jeder, der versucht, die Lagerung, Stabilität, Spezifität, Chemie, Sensitivität oder sogar grundlegende Methoden zur Testvalidierung und/oder -ergebnisse nachzuschlagen, wird durch einen FDA-Bericht blockiert, der lächerlich invasive Schwärzungen enthält, die selbst das grundlegendste wissenschaftliche Verständnis für die potenzielle Bewertung erschweren Ergebnisse oder Durchführung von Tests unmöglich.
Als eindringliches visuelles Beispiel ist eine einzelne redigierte Seite in einer längeren Zusammenfassung der FDA-Zulassungen (siehe unten) Teil eines 127-Seiten-Dokument (davon wurden nur 63 Seiten geteilt und von diesen 63 Seiten wurden etwa 50 % redigiert) zur Bewertung der Reinheit, Konzentration und anderer analytischer Maße von mRNA-Injektionen.
die FDA (b)(4)-Schwärzungen spezifizierte detaillierte Schwärzungen, die verwendet werden, um „Geschäftsgeheimnisse und vertrauliche Geschäfts- oder Finanzinformationen schützen.“ Aber ist es wirklich angemessen, es als „kommerziell“ zu bezeichnen, wenn die Forschung/Entwicklung/das Produkt damit finanziert wurde? Hunderte Millionen Steuergelder?

Ohne eine Liste der Inhaltsstoffe oder Testmethoden ist es für niemanden außer der FDA oder den Herstellern unmöglich, genau zu wissen, wie das Produkt zu prüfen ist Verfälschung (veränderte oder giftige Inhaltsstoffe) oder Fehlbezeichnung (da eine vollständige Liste der Inhaltsstoffe einschließlich der Nukleotidsequenz und der Konfigurationen von Lipid-Nanopartikeln sind auf dem Produktetikett besonders vage).
Der Mangel an Methodik ist besonders problematisch, da neue, vorläufige Daten, die unabhängige Methoden verwenden, Beweise dafür gezeigt haben DNA-Kontamination bei mRNA-Covid-Injektionen.
Wenn also eine externe Person behauptete, eine Verunreinigung in mRNA-Spritzen getestet und festgestellt zu haben, und die FDA oder die Hersteller um eine Antwort bitten würde, erhielt sie eine Antwort, die etwa so aussah:
- Sie haben keine validierte/geeignete Testmethode verwendet, um zu Ihren Schlussfolgerungen zu gelangen, und daher sind Ihre Analysen ungültig.
Dazu würde das unabhängige Labor versuchen, die Testmethodik aus der von der FDA genehmigten Dokumentation anzufordern (d. h. dem vollständigen Dokument, das Folgendes enthält). Abbildung 5) indem Sie fragen: „Okay, ich würde es gerne mit Ihrer genehmigten Methodik testen; verrätst du uns, was das ist?“
- Die FDA oder der Hersteller würden etwa so antworten: „Was wir bereit sind, über die angewandte Methodik offenzulegen, die nicht vertraulich ist, finden Sie online oder über eine FOIA-Anfrage der FDA„... wo man sie treffen würde das folgende stark redigierte Dokument, wo alles, was auch nur annähernd von Bedeutung ist, mit (b)(4)-Schwärzungen überdeckt wird.
Zwischen den Zeilen zu lesen: Es ist offensichtlich, dass weder die Hersteller noch die amerikanische FDA wollen, dass jemand anderes als sie selbst die vollständigen Inhaltsstoffe von mRNA-Injektionen kennt oder diese sogar auf Reinheit und Konsistenz testet.
Laut FDA-Beamten: Pharmazeutische Herstellung ist Höchst Fehleranfällig:
Viele Während des pharmazeutischen Herstellungsprozesses kann und kann etwas schiefgehen. Abgesehen von möglichen Inkonsistenzen bei mRNA/LNP-Injektionen ergeben sich auch qualitative und quantitative Probleme alles, Von der FDA reguliertes Arzneimittel. Sogar das Repräsentantenhaus und der Senat haben Berichte über das Versagen der FDA bei der Sicherung der amerikanischen Arzneimittellieferkette offiziell anerkannt. Die Mehrheit Amerikas Arzneimittel Verbraucher-Endverbraucher-Produkts wird produziert im Ausland in Ländern wie Indien und China, und andere Länder mit niedrigen Arbeitskosten sind es nicht ist für sein hohes Maß an Qualitätskontrolle bekannt. Das Bundesregister ist voller Berichte über Verstöße in indischen und chinesischen Produktionsstätten.
Zertifiziert die FDA diese Anlagen – auch solche mit langen Verstößen in der Vergangenheit – auch über ein „Mail-in“-System an die FDA? Unverschämterweise ist die Antwort auf diese Frage etwas, das jeden, der sich mit pharmazeutischer Qualität beschäftigt, sehr unbehaglich machen würde.
Während Six Sigma Während in der Automobil-, Computer-, Mobiltelefon- und anderen High-Tech-Fertigung das Präzisionsniveau seit langem das Ziel für Qualität und Sicherheit ist, scheint es bei der pharmazeutischen Herstellung größtenteils übersehen worden zu sein.
FDA-Beamte haben Daten veröffentlicht, die eine Ungenauigkeit von 2-3σ (Sigma) bei der pharmazeutischen Herstellung schätzen. Eine 2σ-Qualität entspricht 308,537 Fehler pro 1,000,000 Möglichkeiten. (Bei der pharmazeutischen Herstellung gibt es wahrscheinlich viel mehr als 1,000,000 Fehlermöglichkeiten.) Die FDA ist sich dessen auf höchster Führungsebene bewusst; in der Tat die aktuelle Michael Kopcha, Leiter des Office of Pharmaceutical Quality der FDA hat sogar die obige Six-Sigma-Berechnung geschrieben und veröffentlicht und dabei die Ungenauigkeit der Arzneimittelherstellung beklagt zurück in 2017.
Der Fehlerspielraum für mRNA-Produkte und/oder ihre LNPs könnte gleichmäßig sein weniger präziser als das 2-3σ (je niedriger das σ, desto fehlerhafter ist ein Produkt), da sie Nukleotidmaterial und neuartige LNPs enthalten, was sie wesentlich komplexer als niedermolekulare Arzneimittel macht – ungeachtet ihrer Entwicklung, Herstellung und Freigabe bei „ Warpgeschwindigkeit."
Da selbst die FDA und ihre Beamten eine inhärente Ungenauigkeit bei der Herstellung erkannt haben, warum in der weiten Welt des Sports Erfüllt die FDA ihren Sicherheitsauftrag nicht, indem sie ihre Freigabetests der mRNA-Technologie öffentlich mit der amerikanischen Öffentlichkeit teilt, die sie finanziert?
Schon wieder vor 1862? Sind mRNA-Spritzen die einzigen Medikamente, die die Amerikaner nicht haben? Komplett Informationen zu den Inhaltsstoffen?
Der Mangel an Klarheit über die Anzahl der Sequenzen von mRNA-Aufnahmen und andere wichtige Informationen steht im direkten Gegensatz zu einem anderen von der FDA zugelassenen RNA-basierten Medikament – Patisiran (Onpattro®). Onpattro stellt die Reihenfolge, das Molekulargewicht und die Milligrammstärke seiner Produkte innerhalb der offiziellen FDA transparent zur Verfügung Verpackungskennzeichnung wie in einem Auszug unten dargestellt:


Fehlende Covid-mRNA-Dosisspezifität: 0.3 ml (oder 0.5 ml) von was?
Derzeit liegen uns noch keine grundlegenden Informationen zu den Inhaltsstoffen einer Covid-mRNA-Injektion vor. Apotheker wissen nur, eine bestimmte Menge zu verabreichen Volumen von Flüssigkeit, und das scheinbar ohne Frage. Normalerweise sollte die offizielle Verpackungsetikettierung der FDA die tatsächlichen Inhaltsstoffe in dieser Menge angeben, bei Covid-mRNA-Etiketten jedoch nicht: Sie geben einfach 0.3 ml (oder 0.5 ml) als „Darreichungsform und Stärke“ an.
Darüber hinaus ist, wie Ihnen jeder Gymnasiast sagen kann, 0.3/0.5 ml ein Volumen, kein Stärke. Wir kennen keine quantitativen Einzelheiten darüber, was in diesen 0.3/0.5 ml enthalten ist, wie zum Beispiel: Wie viele LNP-Partikel? Welche Größe/Morphologie dieser LNPs? Wie viele mRNA-Sequenzen enthält dieser Band?




Wird dies von der FDA als ausreichend transparent oder „wahrheitsgemäße Kennzeichnung“ angesehen?
Der oben ausgeschnittene Auszug aus der Packungsbeilage enthält alle Informationen, die die Hersteller den Verbrauchern bezüglich der Dosis – die im Vergleich zu allen anderen FDA-Etiketten völlig unzureichend ist – oder allen, die mehr über die Flüssigkeitsmenge wissen möchten, mitteilen zu injizieren und die 30- oder 100-mcg-Konzentration einer nicht näher bezeichneten mRNA-Sequenz.
Die bemerkenswerte Ungenauigkeit dieser von der FDA zugelassenen Kennzeichnung scheint insbesondere im Widerspruch zu ihrer fast 120 Jahre alten Kennzeichnung zu stehen: „Sie verlangen, dass Lebensmittel und Arzneimittel wahrheitsgemäße Angaben auf der Etikette tragen und bestimmte Reinheits- und Stärkestandards erfüllen"
Ist dies das, was die FDA als „wahrheitsgemäße“ Liste der Inhaltsstoffe bezeichnet? (Sehen 21CFR §352 und 21 CFR §201.10 bezüglich „Inhaltsstoffangaben“ und „falsch gekennzeichnete Arzneimittel und Geräte“.
Die Frage ist: Ist die Auflistung unbekannter oder unspezifischer Inhaltsstoffe möglich, die niemand außer dem Hersteller entschlüsseln kann? wirklich den Geist oder die gesetzlichen Anforderungen der „Kennzeichnung“ erfüllen? Ist diese Bezeichnung das, was die amerikanische FDA als „wahrheitsgemäß“ ansieht? Auf wessen Seite steht überhaupt die FDA; Hersteller oder Verbraucher?
Abgesehen davon, dass es nicht direkt spezifiziert wird, kann die genaue Anzahl der LNP- oder mRNA-Stränge in einer 30- oder 100-mcg-Injektion nicht einmal extrapoliert werden stöchiometrisch oder auf der Grundlage von Avogadros Nummer, da die mRNA-Sequenz, das Molekulargewicht und/oder die LNP-Komponente/-Konfigurationen nirgends in der offiziellen FDA-Kennzeichnung angegeben sind.
Wie kann jemand wissen, ob die Anzahl der mRNA-Stränge, die das Spike-Protein für Covid kodieren, proportional zur Menge an Covid-Inokulum ist, die man durch eine ambulant erworbene Infektion erhalten würde? Antwort: sie können nicht.
Sind Covid-mRNA-Injektionen Entsprechend beschriftet/falsch beschriftet?
21 CFR 211.125 spezifiziert „Die zur Verwendung bei der Etikettierung von Arzneimitteln ausgestellten Etiketten müssen streng kontrolliert werden.Aber es scheint, dass die FDA mit ihrer genehmigten Kennzeichnung von Covid-mRNA-Injektionen trotzdem so lax war Jedes andere Medikament – einschließlich des mRNA-basierten Onpattro – gibt diese Informationen an. Historisch gesehen basieren regulatorische Entscheidungen der FDA (z. B. welche Informationen in die Produktkennzeichnung aufgenommen werden sollen) auf Vorrang, und Covid-mRNA-Impfungen stellten eine offensichtliche Abweichung vom historischen und rechtlichen Vorrang der FDA dar. Dieses bemerkenswerte Fehlen von Daten und die Unklarheit gehen gewissermaßen auf die Zeit zurück Morleys Leber- und Nieren-Cordial im späten 1800. Jahrhundert. Der Unterschied ist: Damals gab es die FDA noch nicht, aber heute gibt es eine FDA mit etwa 20,000 Mitarbeitern, von denen zumindest einige angeblich glaubten, dass diese Kennzeichnung transparent und „wahrheitsgemäß“ sei.
Die Angabe einer unbekannten/nicht entzifferbaren/undurchsichtigen Zutat, deren Wahrscheinlichkeit niemand genau bestimmen konnte, ist nicht das, was die Gesetzgeber des Pure Food and Drug Act von 1906 beabsichtigten, als sie die FDA-Regeln zur „wahrheitsgemäßen Kennzeichnung“ präzisierten. Davon abgesehen: die Tatsache, dass die Dosen pro Volumen bei verschiedenen Herstellern verdoppelt werden (30 µg/0.3 ml vs 100 µg/0.5 ml) bedeutet, dass diese mRNA-Sequenzen in der Nukleotidlänge offenbar stark unterschiedlich sind und dies wiederum auch getan hätte mehr und verschiedene LNPs plus Anhänge. Bedeutet das, dass die zur Transkription des Spike-Proteins verwendeten mRNA-Sequenzen im Vergleich zu anderen Herstellern etwa doppelt so groß sind (10 µg/0.1 ml gegenüber 20 µg/0.1 ml), oder trägt etwas anderes zum Unterschied in der Nukleotidlänge bei?
Für den Laien, der bis hierher noch liest (ein großes Lob übrigens): Das Fehlen detaillierter Kennzeichnungsinformationen könnte so sein, als würde man ein Haus zum Verkauf anbieten, mit der Aussage, es sei aus Holz und Ziegeln, auf einer Betonplatte – aber nicht sichtbar Alle Bilder des Hauses (z. B. Sequenz) und keine Angabe der Quadratmeterzahl (z. B. Molekulargewicht). In jedem Fall ist der Mangel an Informationen unzureichend und eine Abweichung von traditionellen Standards.
Jedes andere von der FDA zugelassene Medikament – einschließlich anderer mRNA-Medikamente – enthält vollständige Angaben zu den Inhaltsstoffen seiner Produkte, einschließlich eine Strukturdarstellung und ein Molekulargewicht ihres Produkts, damit die Leute genau wissen, was sie bekommen.
Es stimmt: Schlagen Sie im Internet nach, welche Droge Ihnen einfällt Drugs.com-Datenbank und beachten Sie, wie alle Markierungen Struktur und/oder Molekulargewicht angeben. Der Beweis, dass Covid-mRNA-Impfungen ein sind auffällig Ausnahme von der historischen FDA-Zulassungspraxis und der „truthful label“-Regel.
Dänische Studie 2023 beschreibt signifikante klinische Variabilität zwischen Chargen von mRNA-Covid-19-mRNA-Injektionen:
Das Fehlen jeglicher Transparenz auch bei der potenziell ungültigen Validierung von per Post eingesandten Tests scheint den Herstellern einen weiteren entscheidenden Teil dessen, was die FDA überwacht, entgangen zu sein: potenzielle klinische Manifestationen bei Chargen-/Chargenvarianten von mRNA-Spritzen. Eine Retrospektive Dänische Sicherheitsstudie In der Anfang 2023 veröffentlichten Studie wurde ein stark abweichendes Muster unerwünschter Ereignisse aus den BNT162b2-mRNA-Injektionen von Pfizer-BioNTech beschrieben, das mit dem dänischen DKMA-Meldesystem für unerwünschte Ereignisse korreliert.
Im folgenden Liniendiagramm stellen verschiedenfarbige Punkte unterschiedliche Chargen der mRNA-Injektionen von Pfizer-BioNTech dar. Die Chargen wurden in drei verschiedene Kategorien unterteilt. hohe, niedrige bis (fast) fehlende Anzahl der gemeldeten unerwünschten Ereignisgruppen (blaue, grüne bzw. gelbe Diagramme).
Mit anderen Worten: Bei vermeintlich „gleichwertigen“ Produkten desselben Herstellers scheinen die Häufigkeit unerwünschter Ereignisse je nach Charge stark zu variieren, wobei jede dieser Chargen Hunderttausende mRNA-Injektionen darstellt.
Als entsprechende lineare Regressionsgeraden hinzugefügt wurden, ergab sich ein besonderes Muster:
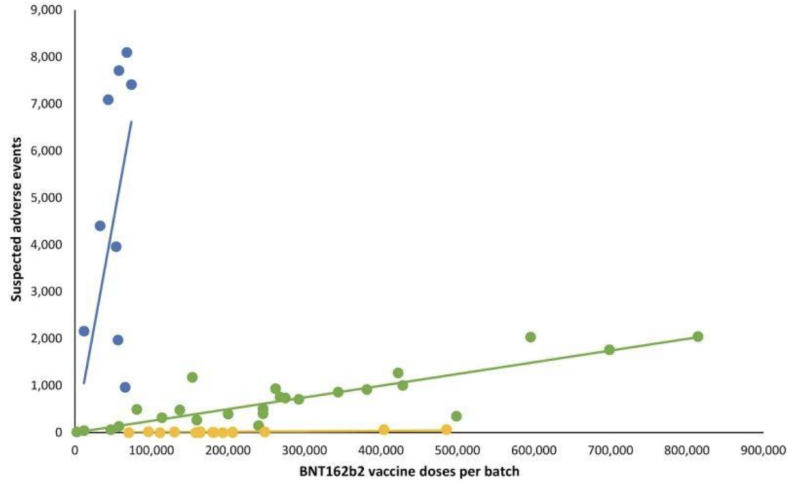
Wichtige Fragen zu den bemerkenswerten Unterschieden bei unerwünschten Ereignissen zwischen Covid-19-mRNA-Chargen sind:
- Könnten Abweichungen bei unerwünschten Ereignissen auf qualitative oder quantitative Abweichungen in den mRNA-Sequenzen oder der Anzahl der mRNA-Stränge zwischen den Chargen zurückzuführen sein?
- Könnten Abweichungen bei unerwünschten Ereignissen auf qualitative oder quantitative Unterschiede in der Größe/Morphologie oder Menge der LNPs zwischen den Chargen zurückzuführen sein? Welche Tests wurden durchgeführt? Gewährleistung der Sicherheit verschiedener LNPs in mRNA-Injektionen verwendet?
- Waren die Chargen, die den gelben gegenüber grünen gegenüber blauen Datenpunkten entsprachen, qualitativ oder quantitativ irgendwie unterschiedlich?
- Wurde die Lagerung/Handhabung nach der Herstellung in der verwaltenden Einrichtung (oder irgendwo anders entlang der Lieferkette) beeinträchtigt, was zu Produktschwankungen führte?
- Wie hoch ist die Sigma-/Fehlerquote dieses und anderer Produkte, die aus der jeweiligen Produktionsstätte/dem jeweiligen Schichtleiter stammen, der für die Fertigung verantwortlich ist?
- Wurden die Inhaltsstoffe dieser Covid-mRNA-Produkte je nach Charge aus Indien oder China und nicht anderswo bezogen?
- Wie viel Prozent der Chargen von Covid-mRNA-Produkten wurden von der Einführung bis heute durch persönliche Entnahme durch einen FDA-Inspektor im Vergleich zu Chargen, die per Post „eingeschickt“ wurden, getestet? Wurde jede einzelne Charge nur mit einer dieser beiden Sammelmethoden getestet?
- Hat die FDA eine Überprüfung der Freigabetests für die Chargen des dänischen DKMA-Meldesystems für unerwünschte Ereignisse durchgeführt? Wenn ja, warum veröffentlicht die FDA diese speziellen Testergebnisse nicht? Wenn nicht, warum wurden keine Tests durchgeführt?
- Gibt es ein grundlegendes Problem bei der zuverlässigen und kontaminationsfreien Produktion von LNPs und/oder mRNA-Sequenzen?
Die Ergebnisse der dänischen Studie und die oben genannten Fragen zu unerwünschten Ereignissen könnten *beginnend* angegangen werden, aber nicht ohne die unabhängige Veröffentlichung der Ergebnisse ihrer Freigabetestergebnisse durch die FDA. Aufgrund der allgegenwärtigen Redaktionen der FDA (b)(4) kennt derzeit niemand die validierte Methodik zum Testen von Covid-mRNA-Impfungen or genau, welche Chargen in der dänischen Studie getestet wurden und welche nicht or die Ergebnisse dieser Chargentests.
… Andererseits: Selbst wenn die FDA beschlossen hätte, diese Chargentestergebnisse zu veröffentlichen, woher wissen Verbraucher dann, ob diese Ergebnisse für die angegebenen Chargen repräsentativ sind, da die Hersteller selbst auswählen, welche Proben sie „einsenden“?
Die fehlende Transparenz der Inhaltsstoffe und die Sicherstellung der Qualität durch eine geeignete Probenahmemethode ist eine grundlegende und grundlegende Anforderung der FDA. Tatsächlich war es der Hauptgrund für die Gründung der FDA! Verdienen die Amerikaner nicht mehr Transparenz, Aufsicht und Gesetze zur „wahrheitsgemäßen Kennzeichnung“ unserer Arzneimittel – insbesondere, da diese Gesetze vor über 100 Jahren erlassen wurden?
Veröffentlicht unter a Creative Commons Namensnennung 4.0 Internationale Lizenz
Für Nachdrucke setzen Sie bitte den kanonischen Link wieder auf das Original zurück Brownstone-Institut Artikel und Autor.








