
Immer mehr prominente Mediziner fordern einen sofortigen Stopp des Einsatzes der angeblich „sicheren und wirksamen“ COVID-19-mRNA-Impfungen. Jetzt hat sich der Generalchirurg des Staates Florida, Dr. Joseph Ladapo, der ständig wachsenden Liste angeschlossen.
Ein Auszug aus der Mitteilung des Gesundheitsministeriums von Florida vom 3. Januar Bekanntmachung lautet:
Der Surgeon General äußerte Bedenken hinsichtlich Nukleinsäurekontaminanten in den zugelassenen COVID-19-mRNA-Impfstoffen von Pfizer und Moderna, insbesondere in Bezug auf das Vorhandensein von Lipid-Nanopartikelkomplexen und Promotor-/Enhancer-DNA des Simian Virus 40 (SV40). Lipid-Nanopartikel sind ein effizientes Vehikel für die Abgabe der mRNA in den COVID-19-Impfstoffen in menschliche Zellen und können daher ein ebenso effizientes Vehikel für die Abgabe kontaminierender DNA in menschliche Zellen sein. Das Vorhandensein von SV40-Promotor-/Enhancer-DNA kann auch ein einzigartiges und erhöhtes Risiko der DNA-Integration in menschliche Zellen darstellen.
Das Bulletin enthielt die folgende Aussage von Dr. Ladapo:
Die DNA-Integration stellt ein einzigartiges und erhöhtes Risiko für die menschliche Gesundheit und die Integrität des menschlichen Genoms dar, einschließlich des Risikos, dass in Spermien oder Eizellen integrierte DNA an Nachkommen von mRNA-COVID-19-Impfstoffempfängern weitergegeben werden könnte. Wenn die Risiken der DNA-Integration für mRNA-COVID-19-Impfstoffe nicht bewertet wurden, sind diese Impfstoffe nicht für die Anwendung beim Menschen geeignet.
Die Warnungen des Florida Surgeon General konzentrierten sich auf die potenziellen Gesundheitsrisiken, die mit der Insertionsmutagenese (der Integration fremder DNA in das Genom des Subjekts/Wirts) verbunden sind. Doch damit nicht genug – er erwähnte noch ein weiteres besorgniserregendes Risiko Generationsmutagenese. Dabei könnte die „in Spermien oder Eizellen integrierte DNA an die Nachkommen von mRNA-COVID-19-Impfstoffempfängern weitergegeben werden.“
Die Grundlage für Dr. Ladapos Warnung hinsichtlich des Vorhandenseins der „SV40-Promotor-/Enhancer-DNA“ in den mRNA-Aufnahmen sind die schockierenden Erkenntnisse einer aktuellen Studie Pre-Print-Studie. Sowohl in den monovalenten als auch in den bivalenten mRNA-Spritzen von Moderna und Pfizer/BioNTech wurden erhebliche Mengen (Milliarden bis Hunderte Milliarden Fragmente von Plasmid-DNA) gefunden, die die behördlichen Standards bei weitem übertrafen. Daraus lässt sich schließen, dass es sich bei den injizierbaren mRNA-Produkten, die in die Arme von Milliarden gelangt sind, um das handelt stark verfälschtsowie die falsche Bezeichnung.
Der Hauptautor der Studie, Molekularvirologe Dr. David J. Speicher, gab mir seine Antwort auf Dr. Ladapos Aussage.
Es ist ermutigend zu sehen, wie mutig Dr. Joseph Ladapo einen Stopp der COVID-19-Impfstoffe fordert, und es ist eine Ehre zu wissen, dass unsere Studie maßgeblich zu dieser Entscheidung beigetragen hat. Unsere kanadische Studie bestätigte frühere Berichte von Kevin McKernan und Dr. Philip Buckhaultz, die hohe Mengen an restlicher Plasmid-DNA in den Lipid-Nanopartikeln in den COVID-19-modRNA-Impfstoffen zeigten. Der SV40-Enhancer-Promotor in den Pfizer-Impfstoffen birgt ein Risiko für die genomische Integration und beeinträchtigt das p53-Gen (den Wächter des Genoms). Beides könnte zu einem erhöhten Krebsrisiko führen. Unsere Daten zur Verfälschung in Verbindung mit dem Nature-Artikel von Mulroney et al., der eine abweichende Immunantwort aufgrund der ribosomalen Rasterverschiebung zeigt, sollten ausreichen, um diese Impfstoffe weltweit zu stoppen. Wir brauchen sofortige Forschung zu den Auswirkungen dieser Impfstoffe auf die menschliche Gesundheit. Ich applaudiere Dr. Ladapo für seine Haltung und hoffe, dass andere Orte, wie Alberta, diesem Beispiel folgen werden.
Im krassen Gegensatz zu Speichers Lob für Ladapo sagte Dr. Paul Offit, Mitglied des Beratungsausschusses für Impfstoffe und verwandte biologische Produkte der Food and Drug Administration (FDA), der für die „Stempelung“ der COVID-19-mRNA-Injektionen verantwortlich ist, in einem Interview: „Es ist kaum zu glauben, dass Dr. Ladapo diese Erklärung tatsächlich abgegeben hat“ und wies anschließend alle Bedenken des Florida Surgeon General zurück.
Einen Monat zuvor, die FDA reagiert auf einen Brief von Dr. Ladapo, in dem es heißt: „Bei über einer Milliarde verabreichten Dosen der mRNA-Impfstoffe wurden keine Sicherheitsbedenken im Zusammenhang mit restlicher DNA festgestellt.“
Der Brief der FDA wurde von Dr. Peter Marks, Direktor des Center for Biologics Evaluation and Research (CBER), verfasst, der Ladapos Behauptungen ebenfalls als „ziemlich unglaubwürdig“ und „irreführend“ zurückwies.
Was die FDA-Beamten jedoch nicht leugnen können, ist, dass es sich bei den mRNA-Spritzen um eine noch experimentellere Form der Gentherapie handelt.
Es handelte sich nie um Impfstoffe
Obwohl sie als „Impfstoffe“ zur Förderung der Aufnahme vermarktet werden, erfüllen sowohl die Moderna- als auch die Pfizer-BioNTech-Impfung die Kriterien einer Gentherapie und gehen darüber hinaus.
In BioNTech's In der Einreichung der US-Sicherheits- und Kommission (SEC) heißt es: „mRNA wird von der FDA als Gentherapieprodukt betrachtet.“ Weiter heißt es: „In dieser neuen potenziellen Kategorie von Therapeutika wurde keine mRNA-Immuntherapie zugelassen und wird möglicherweise auch nie zugelassen. Die Entwicklung von mRNA-Arzneimitteln birgt aufgrund der neuartigen und beispiellosen Natur dieser neuen Kategorie von Therapeutika erhebliche klinische Entwicklungs- und Regulierungsrisiken."
Gentherapie gibt es schon seit mehreren Jahrzehnten und beinhaltet den Einsatz viraler Vektoren, bei denen es sich um modifizierte Viren handelt, die therapeutische Gene in Zielzellen einschleusen. Allerdings sind die mRNA-basierten Impfungen eine völlig „neue und beispiellose“ Klasse für sich.
Frühe Studien haben gezeigt, dass bei der Gentherapie schwerwiegende Gesundheitsrisiken auftreten können, die zu Toxizität, Entzündungen und sogar Krebs führen können. In frühen Gentherapieversuchen gab es bemerkenswerte Fälle von Leukämie, die das Bewusstsein für das Risiko einer Insertionsmutagenese schärften.
Aufgrund des äußerst experimentellen Charakters des injizierbaren mRNA-Produkts würde man annehmen, dass eine strengere und robustere Sicherheitsbewertung durchgeführt worden wäre, bevor es der Öffentlichkeit zugänglich gemacht wurde, aber das war nicht der Fall.
Meine Ermittlungsberichte für Neuigkeiten zum Studienstandort, geschrieben vor fast zwei Jahren, auf dem Fundus von Pfizer/BioNTech-Dokumenten (die die FDA bis 2096 vor der Öffentlichkeit verbergen wollte) enthüllte, dass kritische Sicherheitsstudien in Bezug auf Genotoxizität und Karzinogenität, wurden nie gemachtSie wurden auch nicht als „notwendig“ erachtet, da sie unter dem Deckmantel herkömmlicher Impfstoffe behandelt wurden.
Die neuartigen Inhaltsstoffe sind nicht für den menschlichen Gebrauch bestimmt
Zwei der vier Verbindungen, aus denen die Lipid-Nanopartikel (LNPs) bestehen, die die modifizierte (synthetische) mRNA verkapseln, wurden noch nie zuvor in einem Arzneimittel verwendet: ALC-0315 und ALC-0519, beide lizenziert von Acuitas Therapeutics. Die LNPs von Acuitas sind Bestandteile der mRNA-Covid-19-Impfstoffe von Pfizer/BioNTech und Moderna. Darüber hinaus zeigt die wissenschaftliche Literatur, dass diese LNPs hochgiftig sein können entzündlicher.
Während der Ansprache von Dr. Ryan Cole im britischen Parlamentsbüro, über die ich berichtet habe, teilte er Folgendes mit.
„Im Datenblatt der Lipid-Nanopartikel heißt es, dass diese nicht für den menschlichen oder veterinärmedizinischen Gebrauch bestimmt sind. Diese dienen ausschließlich Forschungszwecken. Dennoch haben sie 5 Milliarden Menschen auf der ganzen Welt getroffen!“
Die vollständige Präsentation von Dr. Cole kann angesehen werden hier.
Das Risiko immunologischer Ereignisse durch abweichende Proteine
Die Aufsichtsbehörde äußerte im Bewertungsbericht der EMA vom Februar 2021 Bedenken hinsichtlich der „verkürzte und modifizierte RNA“ und das Risiko, dass „wenn es in der Zelle vorhanden ist, die Möglichkeit besteht, dass aberrante Proteine wird mit Möglichkeiten für ausgedrückt unerwünschte immunologische Ereignisse"

Interessant ist, dass die EMA dieses Risiko als „gering“ eingestuft hat. Ihre Bedenken, die sie anschließend zurückwiesen, wurden nun jedoch kürzlich in einem bestätigt bahnbrechende Studie zu den Pfizer/BioNTech-mRNA-Aufnahmen der Universität Cambridge. Die Studie zeigte das Auftreten eines „ribosomalen Frameshiftings“, das durch die modifizierte/synthetische mRNA ausgelöst wird, was dazu führt, dass abweichende „unbeabsichtigte Proteine“ exprimiert werden, zusammen mit „unbeabsichtigten“ Immunreaktionen darauf.
Inmitten der unzähligen Warnsignale gegen diese neuartigen „Impfstoffe“ ist eine weitere beunruhigende, aber übersehene Tatsache deutlicher geworden, da neue Beweise ans Licht gekommen sind. Das massenproduzierte mRNA-Produkt, das der Öffentlichkeit vorgestellt wurde, war nicht dasselbe, das in der klinischen Studie von Pfizer getestet wurde. Es sei darauf hingewiesen, dass es die Ergebnisse klinischer Studien waren, die mit weltweitem Tamtam bekannt gegeben wurden und auf denen die Aufsichtsbehörden angeblich ihre Genehmigung beruhten.
Pfizers „Bait-and-Switch“
Vor einigen Monaten habe ich Interview Joshua Guetzkow PhD., der kurz und bündig erklärte, wie Pfizer/BioNTech einen „Köder-und-Schalter“ ihres biologischen Produkts durchführte, das anschließend in die Arme von Milliarden Menschen gespritzt wurde.
Eine wichtige Erkenntnis aus dem Interview war, dass das mRNA-Produkt von Pfizer/BioNTech der breiten Masse zugänglich gemacht wurde nicht dasselbe getestet in den entscheidenden klinischen und nichtklinischen Studien (Tierstudien) von Pfizer.
Dies liegt daran, dass das kommerziell eingeführte Produkt mit einer völlig anderen Methode/einem völlig anderen Verfahren hergestellt wurde. Guetzkow stellte in Bezug auf biologische Produkte kategorisch fest: „Der Prozess ist das Produkt"
Das in der klinischen Studie verwendete Produkt wurde über hergestellt Prozess 1.
- Kleine klinische Chargen, die mit dem teureren PCR-Verfahren hergestellt werden, um die DNA-Matrize zu amplifizieren, die zur Herstellung der modifizierten mRNA für die Impfstoffe verwendet wird.
- Außerdem wurde ein hocheffizienter Filtermechanismus mithilfe von Magnetkügelchen eingesetzt.
Das weltweit verkaufte und vertriebene Produkt wurde unter Verwendung von hergestellt Prozess.
- Großserien werden mit einem viel günstigeren Verfahren hergestellt – E. coli-Bakterien wurde ausgewählt, um die als Matrize für die mRNA verwendete DNA zu replizieren.
- Dies führte zu einer Kontamination sowohl durch die restliche (Plasmid-/Bakterien-)DNA als auch durch die E. coli-Membranen, sogenannte Endotoxine, die ebenfalls stark entzündlich sind.
- Der deutliche Rückgang der RNA-Integrität (ein Maß dafür, wie intakt das RNA-Molekül ist) war eine direkte Folge dieser Umstellung von Prozess 1 auf Prozess 2.
Die durchgesickerten E-Mails der Europäischen Arzneimittel-Agentur
Ich habe ausführlich darüber geschrieben durchgesickerte EMA-E-Mails was erstmals die Tatsache aufdeckte, dass die RNA-Integrität erheblich abnahm. Die private interne E-Mail von Evdokia Korakianiti (einem wissenschaftlichen Administrator der EMA) an seine Kollegen (siehe unten) verdeutlicht diesen signifikanten Rückgang der %RNA-Integrität in den vorgeschlagenen kommerziellen Chargen (hergestellt durch Prozess 2) im Vergleich zu den klinischen Chargen (hergestellt durch Prozess 1). .

Die unten gezeigte durchgesickerte PowerPoint-Präsentation eines Treffens zwischen Pfizer-BioNTech und der EMA vom 26. November 2020 zeigt, wie dieser große Einwand auf schockierende Weise „ausgeräumt“ wurde – die RNA-Integritätsspezifikation wurde einfach herabgesetzt bis 50 %, Das bedeutet, dass bis zur Hälfte aller mRNA-Moleküle in den kommerziellen Chargen verkürzt (nicht intakt) sein durften. Noch wichtiger ist, dass die möglichen Auswirkungen des RNA-Integritätsverlusts im Hinblick auf Sicherheit und Wirksamkeit völlig unbekannt waren.

Die versprochene Vergleichsstudie wurde nie durchgeführt
Als Reaktion auf die Umstellung von Prozess 1 auf Prozess 2, „um einen größeren Produktionsumfang zu unterstützen“, fügte Pfizer eine Änderung zu seinem ursprünglichen Protokoll für klinische Studien hinzu. Sie versprach, eine Vergleichsstudie durchzuführen, um die Sicherheit und Immunogenität bei Personen im Alter von 16 bis 55 Jahren zu untersuchen, die mit „Prozess 1“ und „Prozess 2“ geimpft wurden.

Dieser Protokoll Die Änderung erfolgte am 6. Oktober 2020.

Es sind neue Beweise aufgetaucht, die zeigen, dass nie eine Vergleichsstudie durchgeführt wurde. Auf der Social-Media-Plattform X ein anonymer Account gepostet ein wichtiger Abschnitt aus Pfizers „Protokolländerung 20, 15. September 2022.“
Am 15. September 2022 wurde Pfizer entfernt 'das Ziel ist es, die Sicherheit und Immunogenität von prophylaktischem BNT162b2 bei Personen im Alter von 16 bis 55 Jahren zu beschreiben, wenn sie mit einer Studienintervention geimpft wurden, die durch die Herstellung von „Prozess 1“ oder „Prozess 2“ hergestellt wurde, da die Menge an BNT162b2, die jetzt weltweit unter Verwendung von „Prozess 2“ hergestellt und verabreicht wird, verteilt und verabreicht wird „was den Vergleich ungerechtfertigt macht.“
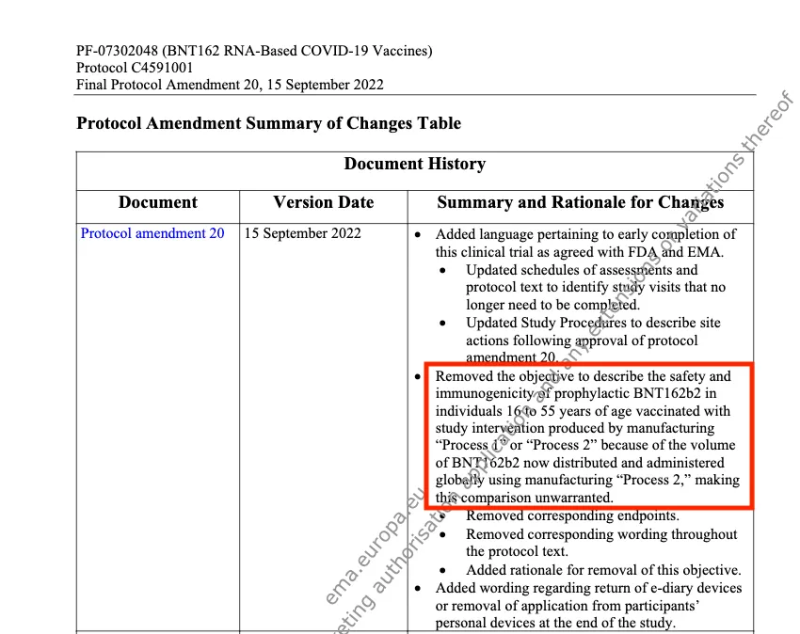
Pfizers Entschuldigung für die Studie lautet: „ungerechtfertigt” weil das Produkt „Prozess 2“ bereits weltweit in großen Mengen eingeführt und verwaltet wurde, ist bedauerlich.
Darüber hinaus lässt sich eine plausible Schlussfolgerung ziehen: Wenn das Produkt „Prozess 1“ dasjenige war, das an Tieren getestet wurde (nichtklinische Studien) und in der klinischen Studie an menschlichen Probanden verwendet wurde, dann ist das Produkt „Prozess 2“, das weltweit eingeführt wurde, Es wurden noch nicht einmal Tierversuche durchgeführt, geschweige denn eine klinische Studie.
Dies geht weit über das Fehlen einer informierten Einwilligung hinaus – wenn die Öffentlichkeit unwissentlich als Laborratten missbraucht wurde.
Wiederveröffentlicht von der Autorin Substack
Veröffentlicht unter a Creative Commons Namensnennung 4.0 Internationale Lizenz
Für Nachdrucke setzen Sie bitte den kanonischen Link wieder auf das Original zurück Brownstone-Institut Artikel und Autor.








