
Die jüngste Befund Die Verbreitung von DNA-Fragmenten in den Covid-19-Impfstoffen von Pfizer und Moderna hat viele zu der Frage veranlasst, warum die FDA, die für die Überwachung der Qualität und Sicherheit der Impfstoffe zuständig ist, es versäumt hat, Alarm zu schlagen.
Die FDA weiß seit Jahren um das Risiko, das von Rest-DNA in Impfstoffen ausgeht. Es ist sein eigenes Orientierungshilfe für die Industrie heißt es:
„Reste DNA könnte aufgrund des onkogenen und/oder infektiösen Potenzials ein Risiko für Ihr Endprodukt darstellen. Es gibt mehrere potenzielle Mechanismen, durch die restliche DNA onkogen sein könnte, einschließlich der Integration und Expression kodierter Onkogene oder der Insertionsmutagenese nach der DNA-Integration.“
Vereinfacht gesagt erkennt die FDA die Möglichkeit an, dass DNA-Fragmente, die beim Herstellungsprozess übrig bleiben, in die eigene DNA eines Patienten eingebaut werden können und möglicherweise Krebs verursachen.
Gemäß den Richtlinien der FDA und der WHO sollte die Menge an Rest-DNA in einer Einzeldosis eines herkömmlichen Impfstoffs 10 ng (ein Milliardstel Gramm) nicht überschreiten.
Aber dieser Grenzwert – der für herkömmliche Impfstoffe verwendet wird – dürfte für die mRNA-Impfstoffe, deren Lipid-Nanopartikel in das Innere der Zellen eindringen können, um die mRNA effizient abzugeben, wahrscheinlich nicht relevant sein.
Eine kürzlich Preprint Artikel von Speicher et al analysierte Chargen der monovalenten und bivalenten mRNA-Impfstoffe in Kanada.
Die Autoren fanden „das Vorhandensein von Milliarden bis Hunderten Milliarden DNA-Molekülen pro Dosis in diesen Impfstoffen.“ Mithilfe der Fluorometrie überschreiten alle Impfstoffe die von der FDA und der WHO festgelegten Richtlinien für Rest-DNA von 10 ng/Dosis.“
Speicher et al berichteten auch über den Fund von DNA-Fragmenten mit mehr als 200 Basenpaaren (ein Maß für die Länge der DNA), was ebenfalls über den FDA-Richtlinien liegt.
Insbesondere bemerkten die Autoren, dass beim Pfizer-Produkt die Rate schwerwiegender unerwünschter Ereignisse umso höher sei, je höher der Anteil an DNA-Fragmenten im Impfstoff sei.
Einige Experten sagen, dass das Risiko einer Genomintegration beim Menschen besteht sehr gering, aber eine neuere Veröffentlichung in Natur fanden heraus, dass etwa 7 Prozent der Zellen integriert werden, wenn sie mit einer Transfektionslösung gemischt werden, die lineare DNA-Stücke enthält.
Ist die FDA besorgt?
Die US-amerikanische Lebensmittel- und Arzneimittelbehörde (FDA) besteht weiterhin darauf, dass jegliche restliche DNA-Kontamination in den Covid-Impfstoffen kein Problem darstellt und dass sie „hinter ihren Erkenntnissen zur Qualität, Sicherheit und Wirksamkeit der mRNA-Impfstoffe steht“.
„Während zuvor Bedenken als theoretische Fragen geäußert wurden, stützen die verfügbaren wissenschaftlichen Beweise die Schlussfolgerung, dass die winzigen Mengen an Rest-DNA weder Krebs noch Veränderungen am genetischen Code einer Person verursachen“, fügte die FDA hinzu.
Die FDA würde nicht die „verfügbaren wissenschaftlichen Beweise“ zur Untermauerung ihrer Behauptung vorlegen, aber es ist erwähnenswert, dass die Produktetiketten der Impfstoffe zeigen, dass Genotoxizitäts- und Karzinogenitätstests durchgeführt wurden nicht vor ihrer Verwendung durchgeführt werden.
David Wiseman, ein forschender Biowissenschaftler, der an der Entwicklung medizinischer Produkte beteiligt ist und Co-Autor der Studie von Speicher et al sagte, die Behauptung der FDA, dass es keine Beweise für einen Krebszusammenhang gebe, werde „unhaltbar“.
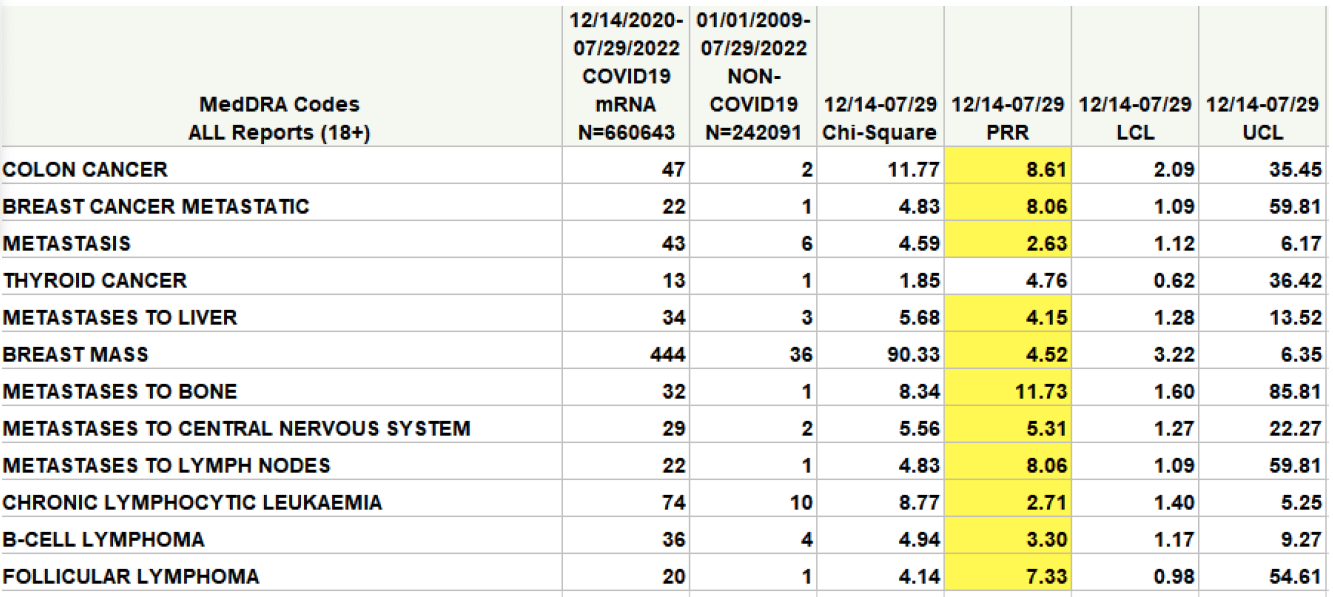
„Die eigene Analyse des CDC zum Sicherheitssignal des Impfstoffs in VAERS zeigt, dass es für einige Krebsarten ein Signal geben könnte“, sagte Wiseman und zeigte auf a berichten Er war Co-Autor und schickte ihn an die Nationalakademien.
In der Tabelle (gelb hervorgehoben) gilt ein Sicherheitssignal als signifikant und einer weiteren Untersuchung wert, wenn der Wert in der mit PRR gekennzeichneten Spalte 2 und der Wert in der Chi-Quadrat-Spalte 4 überschreitet.
Die FDA würde nicht bestätigen, ob sie DNA-Werte gefunden hat, die über akzeptablen Werten liegen, und auch nicht, ob sie weitere Untersuchungen durchführt.
Stattdessen schickte die FDA nach monatelangen Anfragen Standardantworten an mich (und andere Medien), in denen es hieß: „Bei über einer Milliarde verabreichten Dosen der mRNA-Impfstoffe wurden keine Sicherheitsbedenken im Zusammenhang mit restlicher DNA festgestellt.“
Auf eine Liste mit Fragen zu ihren Tests und Aufsicht antwortete die FDA, dass sie „derzeit keine zusätzlichen Informationen zur Verfügung stellen kann“.
Schlechte Fertigungsaufsicht
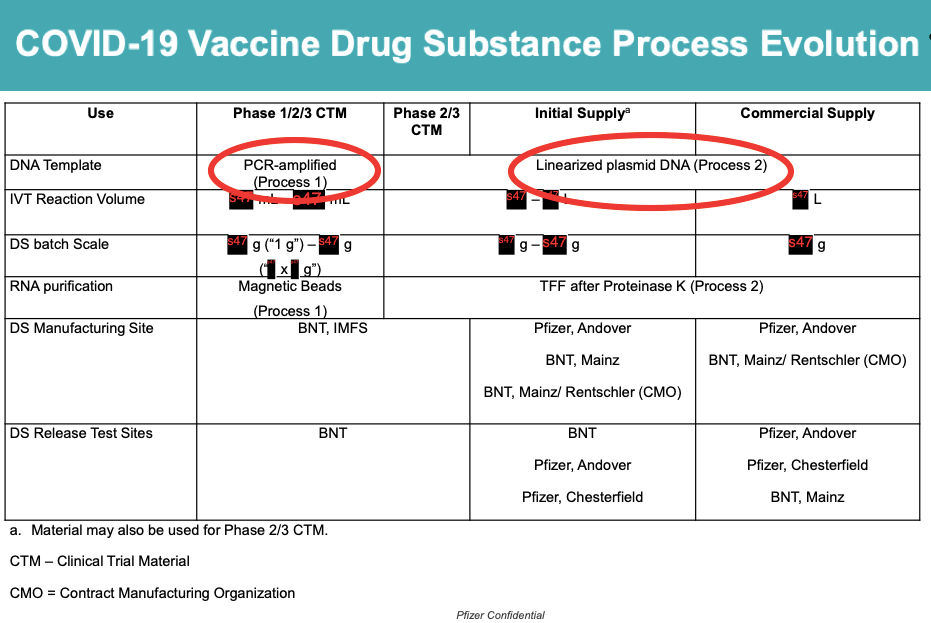
Wir wissen jetzt, dass der Impfstoff von Pfizer in den klinischen Studien (PROZESS 1) verwendet wurde anders hergestellt auf den Impfstoff, der der breiten Bevölkerung injiziert wurde (PROZESS 2).
Dieser wechseln von PROZESS 1 zu PROZESS 2 führte zu Plasmid-DNA-Verunreinigungen (siehe rote Kreise), die das Sicherheitsprofil des Impfstoffs verändern könnten.
Ich fragte die FDA, ob sie über menschliche Daten zum Vergleich der beiden Prozesse verfüge.
Die Behörde verwies mich auf die EUA der FDA Rezensionsnotiz vom 20. November 2020, was darauf hindeutet, dass die Tests „im Gange“ seien.
In dem drei Jahre alten Dokument heißt es: „Eine umfassendere Vergleichbarkeitsbewertung, die zusätzliche Chargen von mehreren DP-Fertigungsknoten umfasst, ist erforderlich.“ laufend und die Ergebnisse werden der EUA nach Abschluss der Studie vorgelegt.“
Als ich die FDA um Zugang zu den „laufenden“ Ergebnissen bat, wurde ich angewiesen, die Informationen von Pfizer einzuholen, aber der Pharmakonzern antwortete nicht auf meine Anfragen.
Eine Informationsfreiheit Anforderung von Nick Hunt von der Täglicher Skeptiker kann erklären, warum.
Pfizer versprach der Aufsichtsbehörde, die Sicherheit und Immunogenität der beiden Prozesse bei den Teilnehmern zu vergleichen und bis Februar 2021 einen Bericht vorzulegen, aber es scheint, dass diese Studien nie durchgeführt wurden.
Das FOI erklärte:
…im Oktober 2020 wurde in der Studie C4591001 ein exploratives Ziel hinzugefügt, um die Sicherheit und Immunogenität von Impfstoffen zu beschreiben, die durch die Herstellung von „Prozess 1“ oder „Prozess 2“ bei Teilnehmern im Alter von 16 bis 55 Jahren hergestellt wurden. Dieses Sondierungsziel wurde im September 20 aufgrund der umfangreichen Verwendung von Impfstoffen, die über „Prozess 2022“ hergestellt wurden, entfernt und in der Protokolländerung 2 dokumentiert. Daher wurde dieser Prozessvergleich nicht im Rahmen der formalen Dokumentation im Rahmen der Protokolländerung durchgeführt.[Betonung hinzugefügt]
Wiseman sagte: „Angesichts des Ausmaßes der Prozessänderung hätte man aufgrund meiner Erfahrung in der Entwicklung medizinischer Produkte sicherlich erwarten können, dass Pfizer solche Studien zur biologischen Vergleichbarkeit durchführt.“
Er fügte hinzu: „Die Tatsache, dass Pfizer eine Freikarte erhalten hat, deutet auf einen erheblichen Mangel an regulatorischer Aufsicht hin.“
Kevin McKernan, der Genomexperte, der Anfang des Jahres DNA-Fragmente in den Impfstoffen entdeckte, sagt, es gebe für Pfizer derzeit „keinen Anreiz“, diese Vergleichstests durchzuführen.
„Das sind Spekulationen meinerseits, aber ich vermute, dass sie es gesehen haben könnten erhöhte unerwünschte Ereignisse „Wir haben die Daten mit der kommerziellen Charge gelöscht und die Daten vergraben, obwohl wir wussten, dass der Zug zu diesem Zeitpunkt den Bahnhof verlassen hatte“, sagte McKernan.
„Es gab keinen politischen Willen, die Impfung zu stoppen, und Pfizer wusste wahrscheinlich, dass die Aufsichtsbehörden es ihnen erlauben würden, die kommerziellen Chargen nicht für die Bevölkerung zu testen“, fügte er hinzu.
Wiederveröffentlicht von der Autorin Substack
Veröffentlicht unter a Creative Commons Namensnennung 4.0 Internationale Lizenz
Für Nachdrucke setzen Sie bitte den kanonischen Link wieder auf das Original zurück Brownstone-Institut Artikel und Autor.








